Revatio® 10 mg/ml Pulver zur Herstellung einer Suspension zum Einnehmen
Nach Rekonstitution enthält jeder ml der Suspension zum Einnehmen 10 mg Sildenafil (als Citrat).
Eine Flasche (112 ml) der rekonstituierten Suspension zum Einnehmen enthält 1,12 g Sildenafil (als Citrat).
Sonstige Bestandteile mit bekannter Wirkung
Jeder Milliliter der rekonstituierten Suspension zum Einnehmen enthält 250 mg Sorbitol (Ph.Eur.).
Jeder Milliliter der rekonstituierten Suspension zum Einnehmen enthält 1 mg Natriumbenzoat.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
Pulver zur Herstellung einer Suspension zum Einnehmen
Weißes bis weißliches Pulver
Erwachsene
Behandlung von erwachsenen Patienten mit pulmonaler arterieller Hypertonie (PAH) der WHO-Funktionsklassen II und III zur Verbesserung der körperlichen Leistungsfähigkeit. Die Wirksamkeit konnte nachgewiesen werden bei primärer PAH und bei pulmonaler Hypertonie in Verbindung mit einer Bindegewebskrankheit.
Kinder und Jugendliche
Behandlung von pädiatrischen Patienten im Alter von 1 bis 17 Jahren mit pulmonaler arterieller Hypertonie. Die Wirksamkeit konnte anhand der Verbesserung der körperlichen Belastbarkeit oder der pulmonalen Hämodynamik nachgewiesen werden bei primärer PAH und bei pulmonaler Hypertonie in Verbindung mit angeborenen Herzerkrankungen (siehe Abschnitt 5.1).
Die Behandlung sollte nur durch einen Arzt eingeleitet und überwacht werden, der Erfahrung mit der Behandlung von PAH hat. Im Falle einer klinischen Verschlechterung trotz einer Behandlung mit Revatio sollten andere Formen der Behandlung in Erwägung gezogen werden.
Dosierung
Erwachsene
Die empfohlene Dosierung beträgt 20 mg dreimal täglich. Der Arzt sollte den Patienten, der eine Einnahme von Revatio vergessen hat, anhalten, so bald wie möglich eine Dosis einzunehmen und dann mit der normalen Dosierung fortzufahren. Zum Ausgleichen einer vergessenen Einnahme sollten die Patienten keine doppelte Dosis einnehmen.
Kinder und Jugendliche (1 bis 17 Jahre)
Bei pädiatrischen Patienten im Alter von 1 Jahr bis 17 Jahren beträgt die empfohlene Dosierung bei einem Körpergewicht ≤ 20 kg 10 mg (1 ml zubereitete Suspension) dreimal täglich und bei einem Körpergewicht > 20 kg 20 mg (2 ml zubereitete Suspension) dreimal täglich. Höhere als die empfohlenen Dosen sollten bei pädiatrischen Patienten mit PAH nicht angewendet werden (siehe Abschnitte 4.4 und 5.1).
Patienten, die zusätzlich weitere Arzneimittel anwenden
Generell sollte jede Dosisanpassung nur nach einer sorgfältigen Nutzen-Risiko-Abschätzung vorgenommen werden. Wenn Sildenafil Patienten verabreicht wird, die bereits CYP3A4-Hemmer wie Erythromycin oder Saquinavir erhalten, sollte eine Dosisreduktion auf zweimal täglich 20 mg erwogen werden. Bei gleichzeitiger Gabe mit stärkeren CYP3A4-Hemmern wie Clarithromycin, Telithromycin und Nefazodon wird eine Dosisreduktion auf einmal täglich 20 mg empfohlen. Zur gleichzeitigen Anwendung von Sildenafil mit den stärksten CYP3A4-Hemmern siehe Abschnitt 4.3. Bei gleichzeitiger Gabe mit CYP3A4-Induktoren kann eine Dosisanpassung für Sildenafil notwendig werden (siehe Abschnitt 4.5).
Spezielle Patientenpopulationen
Ältere Patienten (≥ 65 Jahre)
Bei älteren Patienten ist keine Dosisanpassung erforderlich. Die anhand der 6-Minuten-Gehstrecke gemessene klinische Wirksamkeit kann bei älteren Patienten verringert sein.
Eingeschränkte Nierenfunktion
Bei Patienten mit eingeschränkter Nierenfunktion einschließlich solchen mit schwerer Niereninsuffizienz (Kreatinin-Clearance < 30 ml/min) ist keine initiale Dosisanpassung erforderlich. Nur wenn die Therapie nicht gut vertragen wird, sollte nach einer sorgfältigen Nutzen-Risiko-Bewertung eine Dosisreduktion auf 20 mg zweimal täglich erwogen werden.
Eingeschränkte Leberfunktion
Bei Patienten mit eingeschränkter Leberfunktion (Child-Pugh-Klassen A und B) ist keine initiale Dosisanpassung erforderlich. Nur wenn die Therapie nicht gut vertragen wird, sollte nach einer sorgfältigen Nutzen-Risiko-Bewertung eine Dosisreduktion auf 20 mg zweimal täglich erwogen werden.
Bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse C) ist Revatio kontraindiziert (siehe Abschnitt 4.3).
Kinder und Jugendliche (Kinder unter 1 Jahr und Neugeborene)
Außerhalb der zugelassenen Indikationen darf Sildenafil nicht bei Neugeborenen mit persistierender pulmonaler Hypertonie des Neugeborenen (PPHN) angewendet werden, da das Risiko größer als der Nutzen ist (siehe Abschnitt 5.1). Die Sicherheit und Wirksamkeit von Revatio bei Kindern unter 1 Jahr mit anderen Erkrankungen sind nicht erwiesen. Es liegen keine Daten vor.
Absetzen der Behandlung
Anhand bisheriger, limitierter Daten ist anzunehmen, dass ein plötzliches Absetzen von Revatio keinen Rebound-Effekt mit einer Verschlechterung der PAH verursacht. Allerdings sollte zur Vermeidung einer möglichen und plötzlichen klinischen Verschlechterung während des Absetzens eine allmähliche Dosisreduktion in Erwägung gezogen werden. Während des Absetzens wird eine engmaschigere Überwachung empfohlen.
Art der Anwendung
Revatio Pulver zur Herstellung einer Suspension zum Einnehmen ist nur zum Einnehmen bestimmt. Die zubereitete Suspension zum Einnehmen (eine weiße Suspension zum Einnehmen mit Traubengeschmack) sollte in Abständen von etwa 6 bis 8 Stunden mit oder unabhängig von einer Mahlzeit eingenommen werden.
Schütteln Sie die Flasche sorgfältig für mindestens 10 Sekunden, bevor Sie die benötigte Dosis entnehmen.
Anleitung zur Zubereitung des Arzneimittels vor der Verabreichung siehe Abschnitt 6.6.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile
Gleichzeitige Gabe mit NO-Donatoren (wie Amylnitrit) oder Nitraten jeglicher Form aufgrund der hypotensiven Effekte von Nitraten (siehe Abschnitt 5.1)
Die Begleittherapie von PDE5-Hemmern, inklusive Sildenafil, mit Guanylatcyclase-Stimulatoren wie Riociguat ist kontraindiziert, da es möglicherweise zu einer symptomatischen Hypotonie kommen kann (siehe Abschnitt 4.5).
Kombination mit den stärksten CYP3A4-Hemmern (z. B. Ketoconazol, Itraconazol, Ritonavir) (siehe Abschnitt 4.5)
Patienten, die aufgrund einer nicht arteriitischen anterioren ischämischen Optikusneuropathie (NAION) ihre Sehkraft auf einem Auge verloren haben, unabhängig davon, ob der Sehverlust mit einer vorherigen Einnahme eines PDE5-Hemmers in Zusammenhang stand oder nicht (siehe Abschnitt 4.4)
Die Sicherheit von Sildenafil wurde bei folgenden Patientengruppen nicht untersucht und seine Anwendung bei diesen Gruppen ist daher kontraindiziert:
schwere Einschränkung der Leberfunktion
kürzlich zurückliegender Schlaganfall oder Herzinfarkt
ausgeprägte Hypotonie (Blutdruck < 90/50 mmHg) bei Behandlungsbeginn
Die Wirksamkeit von Revatio bei Patienten mit schwerer PAH (Funktionsklasse IV) wurde bisher nicht untersucht. Falls sich der klinische Zustand verschlechtert, sollten Therapien in Erwägung gezogen werden, die für ein schweres Stadium der Krankheit empfohlen werden (z. B. Epoprostenol) (siehe Abschnitt 4.2). Das Nutzen-Risiko-Profil von Sildenafil bei Patienten mit PAH der WHO-Funktionsklasse I wurde nicht untersucht.
Studien mit Sildenafil wurden bei PAH in Verbindung mit primären (idiopathischen) Bindegewebskrankheiten und mit angeborenen Herzerkrankungen durchgeführt (siehe Abschnitt 5.1). Die Anwendung von Sildenafil bei anderen Formen der PAH wird nicht empfohlen.
In der pädiatrischen Langzeit-Anschlussstudie wurde bei Patienten, die höhere als die empfohlenen Dosen erhielten, eine Zunahme von Todesfällen beobachtet. Deshalb sollten bei pädiatrischen Patienten mit PAH höhere als die empfohlenen Dosen nicht angewendet werden (siehe auch Abschnitte 4.2 und 5.1).
Retinitis pigmentosa
Die Sicherheit von Sildenafil wurde bei Patienten mit bekannter erblich bedingter degenerativer Retinaerkrankung wie beispielsweise Retinitis pigmentosa (eine Minderheit dieser Patienten hat eine genetisch bedingte Störung der retinalen Phosphodiesterasen) nicht untersucht und seine Anwendung kann daher nicht empfohlen werden.
Gefäßerweiternde Wirkung
Bei der Verordnung von Sildenafil sollen Ärzte sorgfältig abwägen, ob Patienten mit bestimmten Grunderkrankungen durch die leichte bis mäßige gefäßerweiternde Wirkung von Sildenafil beeinträchtigt werden könnten. Hierzu zählen beispielsweise Patienten mit Hypotonie, solche mit Dehydratation, Patienten mit einer schweren Obstruktion des linksventrikulären Ausflusstrakts oder schwerer Einschränkung der autonomen Blutdruckkontrolle (siehe Abschnitt 4.4).
Kardiovaskuläre Risikofaktoren
Nach der Markteinführung von Sildenafil zur Behandlung der erektilen Dysfunktion wurden schwerwiegende kardiovaskuläre Ereignisse, einschließlich Herzinfarkt, instabiler Angina pectoris, plötzlichen Herztodes, ventrikulärer Arrhythmie, zerebrovaskulärer Blutung, transitorischer ischämischer Attacke, Hypertonie und Hypotonie im zeitlichen Zusammenhang mit der Anwendung von Sildenafil berichtet. Die meisten dieser Patienten hatten vorbestehende kardiovaskuläre Risikofaktoren. Für viele Ereignisse wurde berichtet, dass sie während oder kurz nach dem Geschlechtsverkehr auftraten, und für einige wenige, dass sie kurz nach der Anwendung von Sildenafil ohne sexuelle Aktivität auftraten. Es ist nicht möglich zu beurteilen, ob diese Ereignisse direkt mit den kardiovaskulären Risikofaktoren oder mit anderen Faktoren in Zusammenhang stehen.
Priapismus
Eine Anwendung von Sildenafil bei Patienten mit anatomischen Penismissbildungen (wie etwa Angulation, Fibrose im Bereich der Corpora cavernosa oder die Peyronie-Krankheit) sowie bei Patienten mit für Priapismus prädisponierenden Erkrankungen (wie Sichelzellenanämie, Plasmozytom, Leukämie) sollte mit entsprechender Vorsicht erfolgen.
Nach der Markteinführung wurden verlängerte Erektionen und Priapismus unter Sildenafil-Behandlung berichtet. Falls eine Erektion länger als 4 Stunden anhält, sollte der Patient sofort medizinische Hilfe suchen. Wird ein Priapismus nicht sofort behandelt, kann dies zu Gewebeschädigung im Penis und dauerhaftem Potenzverlust führen (siehe Abschnitt 4.8).
Vaso-okklusive Krise bei Patienten mit Sichelzellenanämie
Bei Patienten mit pulmonaler Hypertonie nach einer Sichelzellenanämie sollte Sildenafil nicht angewendet werden. In einer klinischen Studie wurden vaso-okklusive Krisen, die zu einer Krankenhauseinweisung führten, bei den Patienten, die Revatio erhielten, häufiger berichtet als unter Placebo, was zu einem vorzeitigen Abbruch dieser Studie führte.
Sehstörungen
In Zusammenhang mit der Einnahme von Sildenafil und anderen PDE5-Hemmern sind Fälle von Sehstörungen spontan berichtet worden. Fälle von nicht arteriitischer anteriorer ischämischer Optikusneuropathie, einer seltenen Erkrankung, sind in Zusammenhang mit der Einnahme von Sildenafil und anderen PDE5-Hemmern spontan und in einer Beobachtungsstudie berichtet worden (siehe Abschnitt 4.8). Im Falle jeglicher plötzlicher Sehstörungen sollte die Behandlung sofort abgebrochen und eine alternative Therapie in Betracht gezogen werden (siehe Abschnitt 4.3).
Alphablocker
Wenn Patienten unter Alphablocker-Therapie Sildenafil erhalten, ist Vorsicht geboten, da eine gleichzeitige Anwendung bei empfindlichen Personen zu symptomatischer Hypotonie führen kann (siehe Abschnitt 4.5). Um die Möglichkeit einer orthostatischen Hypotonie möglichst gering zu halten, sollten Patienten, die mit Alphablockern behandelt werden, vor Beginn der Behandlung mit Sildenafil hämodynamisch stabil eingestellt sein. Ärzte sollten die Patienten darüber aufklären, wie sie sich beim Auftreten von Symptomen einer orthostatischen Hypotonie verhalten sollen.
Blutungsstörungen
Studien an menschlichen Thrombozyten erbrachten Hinweise, dass Sildenafil die antiaggregatorische Wirkung von Nitroprussid-Natrium in vitro verstärkt. Es liegen keine Daten über die Unbedenklichkeit der Anwendung von Sildenafil an Patienten mit Blutungsstörungen oder aktiven peptischen Ulzera vor. Daher sollte die Gabe von Sildenafil bei diesen Patienten nur nach sorgfältiger Nutzen-Risiko-Abwägung erfolgen.
Vitamin-K-Antagonisten
Bei Patienten mit PAH, insbesondere bei einer PAH in Verbindung mit einer Bindegewebskrankheit, besteht bei Beginn einer Therapie mit Sildenafil unter laufender Behandlung mit einem Vitamin-K-Antagonisten möglicherweise ein erhöhtes Blutungsrisiko.
Venenverschlusskrankheit
Für die Anwendung von Sildenafil bei Patienten mit pulmonaler Hypertonie in Verbindung mit einer pulmonalen Venenverschlusskrankheit liegen bisher keine Daten vor. Allerdings wurden für die Anwendung von Vasodilatatoren (hauptsächlich Prostacyclin) bei solchen Patienten Fälle von lebensbedrohlichen Lungenödemen beschrieben. Sollten daher bei der Anwendung von Sildenafil bei Patienten mit pulmonaler Hypertonie Zeichen eines Lungenödems auftreten, ist an die Möglichkeit einer Venenverschlusskrankheit zu denken.
Sonstige Bestandteile
Revatio 10 mg/ml Pulver zur Herstellung einer Suspension zum Einnehmen enthält Sorbitol (Ph.Eur.), welches eine Quelle für Fructose ist. Patienten mit der seltenen hereditären Fructose-Intoleranz (HFI) dürfen dieses Arzneimittel nicht einnehmen.
Revatio 10 mg/ml Pulver zur Herstellung einer Suspension zum Einnehmen enthält 1 mg Natriumbenzoat pro Milliliter rekonstituierter Suspension zum Einnehmen. Eine Zunahme des Bilirubingehalts im Blut nachVerdrängung von Albumin kann einen Neugeborenenikterus verstärken und zu einem Kernikterus (nicht-konjugierte Bilirubinablagerungen im Hirngewebe) führen.
Revatio 10 mg/ml Pulver zur Herstellung einer Suspension zum Einnehmen enthält weniger als 1 mmol (23 mg) Natrium pro Milliliter rekonstituierter Suspension zum Einnehmen. Patienten unter einer natriumarmen Diät können darüber informiert werden, dass dieses Arzneimittel nahezu „natriumfrei“ ist.
Anwendung von Sildenafil zusammen mit Bosentan
Die Wirksamkeit von Sildenafil bei Patienten, die gleichzeitig Bosentan erhalten, wurde nicht abschließend nachgewiesen (siehe Abschnitte 4.5 und 5.1).
Gleichzeitige Gabe von anderen PDE5-Hemmern
Die Sicherheit und Wirksamkeit von Sildenafil in Kombination mit anderen PDE5-Hemmern, einschließlich Viagra, sind bei PAH-Patienten nicht untersucht worden. Die Anwendung in dieser Kombination wird nicht empfohlen (siehe Abschnitt 4.5).
Wirkungen anderer Arzneimittel auf Sildenafil
In-vitro-Studien
Der Sildenafil-Metabolismus wird grundsätzlich durch die Cytochrom-P450 (CYP)-Isoenzyme 3A4 (Hauptweg) und 2C9 (Nebenweg) vermittelt. Die Sildenafil-Clearance kann folglich durch Inhibitoren dieser Isoenzyme herabgesetzt und durch Induktoren dieser Enzyme erhöht sein. Zu Dosisempfehlungen siehe Abschnitte 4.2 und 4.3.
In-vivo-Studien
Die gleichzeitige Gabe von oral appliziertem Sildenafil und intravenös appliziertem Epoprostenol wurde untersucht (siehe Abschnitte 4.8 und 5.1).
Die Wirksamkeit und Sicherheit bei gleichzeitiger Gabe von Sildenafil mit anderen Behandlungen der PAH (z. B. Ambrisentan, Iloprost) wurden in kontrollierten klinischen Studien nicht untersucht. Daher ist im Falle gleichzeitiger Gabe Vorsicht geboten.
Die Sicherheit und Wirksamkeit von Sildenafil bei gleichzeitiger Gabe mit anderen PDE5-Inhibitoren wurden bei Patienten mit PAH nicht untersucht (siehe Abschnitt 4.4).
Eine populationspharmakokinetische Analyse der Daten für alle Patientengruppen in klinischen Studien bei PAH zeigte eine reduzierte Sildenafil-Clearance und/oder eine erhöhte orale Bioverfügbarkeit von Sildenafil bei gemeinsamer Anwendung mit CYP3A4-Substraten und mit der Kombination von CYP3A4-Substraten und Betablockern. Diese waren die einzigen Faktoren mit einem statistisch signifikanten Einfluss auf die Pharmakokinetik von Sildenafil bei Patienten mit PAH. Die Plasma-AUC von Sildenafil war bei Patienten mit CYP3A4-Substraten und CYP3A4-Substraten plus Betablockern um 43 % bzw. 66 % höher als bei Patienten, die keine solchen Arzneimittel erhielten. Die Plasma-AUC von Sildenafil war bei einer Dosis von 80 mg dreimal täglich um das 5-Fache höher als bei einer Dosis von 20 mg dreimal täglich. Dieser Konzentrationsbereich entspricht der Erhöhung der Sildenafil-Exposition, die bei speziell konzipierten Interaktionsstudien mit CYP3A4-Hemmern (mit Ausnahme der stärksten CYP3A4-Hemmer wie Ketoconazol, Itraconazol, Ritonavir) beobachtet wurden.
CYP3A4-Induktoren dürften einen erheblichen Einfluss auf die Pharmakokinetik von Sildenafil bei Patienten mit PAH haben. Dies konnte in einer In-vivo-Interaktionsstudie mit dem CYP3A4-Induktor Bosentan bestätigt werden.
Bei gesunden Freiwilligen führte die gleichzeitige Anwendung von zweimal täglich 125 mg Bosentan (einem mäßigen Induktor von CYP3A4, CYP2C9 und möglicherweise auch von CYP2C19) mit dreimal täglich 80 mg Sildenafil (im Steady State) über 6 Tage zu einer Verringerung der AUC von Sildenafil um 63 %. Bei einer populationspharmakokinetischen Analyse von Sildenafil-Daten erwachsener PAH-Patienten in klinischen Studien, darunter eine 12-wöchige Studie zur Beurteilung der Wirksamkeit und Sicherheit von 20 mg Sildenafil oral dreimal täglich zusätzlich zu einer stabilen Dosis von Bosentan (62,5 mg bis 125 mg zweimal täglich), zeigte sich eine Verminderung der Sildenafil-Exposition unter der gleichzeitigen Gabe von Bosentan, ähnlich wie sie bei gesunden Freiwilligen beobachtet wurde (siehe Abschnitte 4.4 und 5.1).
Bei Patienten, die gleichzeitig starke CYP3A4-Induktoren wie Carbamazepin, Phenytoin, Phenobarbital, Johanniskraut und Rifampicin anwenden, muss die Wirksamkeit von Sildenafil genau überwacht werden.
Die gleichzeitige Anwendung des HIV-Protease-Hemmers Ritonavir, eines hochpotenten P450-Hemmstoffs, im Steady State (zweimal täglich 500 mg) mit Sildenafil (100-mg-Einzeldosis) bewirkte eine 300%ige (4‑fache) Steigerung der Cmax von Sildenafil und eine 1.000%ige (11‑fache) Steigerung der Plasma-AUC von Sildenafil. Nach 24 Stunden betrugen die Sildenafil-Plasmaspiegel noch immer etwa 200 ng/ml im Vergleich zu etwa 5 ng/ml nach alleiniger Gabe von Sildenafil. Dies entspricht den ausgeprägten Effekten von Ritonavir auf ein breites Spektrum von P450-Substraten. Aufgrund dieser pharmakokinetischen Ergebnisse ist die gleichzeitige Einnahme von Sildenafil und Ritonavir bei Patienten mit PAH kontraindiziert (siehe Abschnitt 4.3).
Die gleichzeitige Anwendung des HIV-Protease-Hemmers Saquinavir, eines CYP3A4-Hemmstoffs, im Steady State (dreimal täglich 1.200 mg) mit Sildenafil (100-mg-Einzeldosis) bewirkte eine 140%ige Steigerung der Cmax von Sildenafil und eine 210%ige Steigerung der Plasma-AUC von Sildenafil. Sildenafil hatte keine Auswirkungen auf die Pharmakokinetik von Saquinavir. Zu Dosisempfehlungen siehe Abschnitt 4.2.
Bei Anwendung einer Einzeldosis von 100 mg Sildenafil gemeinsam mit Erythromycin, einem mäßigen CYP3A4-Hemmstoff, im Steady State (zweimal täglich 500 mg für 5 Tage) erhöhte sich die systemische Sildenafil-Exposition (AUC) um 182 %. Zu Dosisempfehlungen siehe Abschnitt 4.2. Bei gesunden männlichen Freiwilligen konnte kein Einfluss von Azithromycin (500 mg täglich über 3 Tage) auf die AUC, Cmax, tmax, Eliminationsrate oder auf die sich daraus ergebende Halbwertszeit von Sildenafil oder auf seinen Hauptmetaboliten im Kreislauf festgestellt werden. Eine Dosisanpassung ist nicht notwendig. Cimetidin (800 mg), ein Cytochrom-P450-Hemmstoff und ein unspezifischer CYP3A4-Hemmstoff, bewirkte eine 56%ige Steigerung der Sildenafil-Plasmaspiegel, wenn es gesunden Freiwilligen gleichzeitig mit Sildenafil (50 mg) gegeben wurde. Eine Dosisanpassung ist nicht notwendig.
Bei den stärksten CYP3A4-Hemmern wie beispielsweise Ketoconazol oder Itraconazol dürften ähnliche Effekte wie bei Ritonavir zu erwarten sein (siehe Abschnitt 4.3). Bei CYP3A4-Hemmern wie z. B. Clarithromycin, Telithromycin und Nefazodon wird erwartet, dass der Effekt zwischen dem von Ritonavir und dem von CYP3A4-Hemmern wie z. B. Saquinavir oder Erythromycin liegt – man vermutet eine 7‑fach höhere Exposition. Bei CYP3A4-Hemmern werden daher Dosisanpassungen empfohlen (siehe Abschnitt 4.2).
Eine populationspharmakokinetische Analyse der Daten für alle Patientengruppen mit PAH lässt vermuten, dass die gemeinsame Anwendung von Betablockern mit CYP3A4-Substraten zu einer zusätzlichen Erhöhung der Plasma-AUC von Sildenafil im Vergleich zu einer alleinigen Anwendung des CYP3A4-Substrats führen könnte.
Grapefruitsaft ist ein schwacher Hemmstoff des CYP3A4-Stoffwechsels in der Darmwand und kann eine geringe Steigerung der Sildenafil-Plasmaspiegel bewirken. Eine Dosisanpassung ist nicht notwendig, die gleichzeitige Einnahme von Sildenafil mit Grapefruitsaft wird jedoch nicht empfohlen.
Durch die Einmalgabe eines Antazidums (Magnesiumhydroxid/Aluminiumhydroxid) wurde die Bioverfügbarkeit von Sildenafil nicht beeinflusst.
Die gleichzeitige Anwendung von oralen Kontrazeptiva (Ethinylestradiol 30 µg und Levonorgestrel 150 µg) hatte keinen Einfluss auf die Pharmakokinetik von Sildenafil.
Nicorandil ist ein Wirkstoff, der gleichzeitig die Kaliumkanäle aktiviert und als Nitrat wirkt. Aufgrund der Nitratkomponente besteht die Möglichkeit einer schwerwiegenden Wechselwirkung mit Sildenafil (siehe Abschnitt 4.3).
Wirkungen von Sildenafil auf andere Arzneimittel
In-vitro-Studien
Sildenafil ist ein schwacher Inhibitor der Cytochrom-P450-Isoenzyme 1A2, 2C9, 2C19, 2D6, 2E1 und 3A4 (IC50 > 150 µM).
Es liegen keine Daten hinsichtlich Wechselwirkungen zwischen Sildenafil und unspezifischen Phosphodiesteraseinhibitoren wie Theophyllin oder Dipyridamol vor.
In-vivo-Studien
Bei gleichzeitiger Anwendung von Sildenafil (50 mg) zeigten sich keine signifikanten Wechselwirkungen mit Tolbutamid (250 mg) oder mit Warfarin (40 mg), die beide durch CYP2C9 verstoffwechselt werden.
Sildenafil hatte keine signifikante Wirkung auf die Plasma-AUC von Atorvastatin (AUC um 11 % erhöht), was vermuten lässt, dass Sildenafil keinen klinisch relevanten Effekt auf CYP3A4 hat.
Es wurden keine Wechselwirkungen zwischen Sildenafil (100-mg-Einzeldosis) und Acenocoumarol beobachtet.
Die durch Acetylsalicylsäure (150 mg) bewirkte Verlängerung der Blutungszeit wurde durch Sildenafil (50 mg) nicht gesteigert.
Die blutdrucksenkende Wirkung von Alkohol (maximale Blutalkoholspiegel im Mittel 80 mg/dl) wurde bei gesunden Probanden durch Sildenafil (50 mg) nicht verstärkt.
In einer Studie an gesunden Freiwilligen führte Sildenafil im Steady State (80 mg dreimal täglich) zu einer Erhöhung der AUC von Bosentan (125 mg zweimal täglich) um 50 %. Eine populationspharmakokinetische Analyse der Daten aus einer Studie mit erwachsenen PAH-Patienten mit einer bestehenden Bosentan-Therapie (62,5 mg bis 125 mg zweimal täglich) ergab bei gleichzeitiger Gabe von Sildenafil im Steady State (20 mg dreimal täglich) eine Erhöhung (20 % [KI 95 %: 9,8 bis 30,8]) der AUC von Bosentan, die geringer war als die bei gesunden Freiwilligen mit gleichzeitiger Gabe von 80 mg Sildenafil dreimal täglich (siehe Abschnitte 4.4 und 5.1).
In einer Interaktionsstudie erhielten Hypertoniker Sildenafil (100 mg) zusammen mit Amlodipin. Es zeigte sich eine zusätzliche Senkung des Blutdrucks im Liegen um 8 mmHg systolisch und um 7 mmHg diastolisch. Das Ausmaß dieser zusätzlichen Blutdrucksenkung war ähnlich der Blutdrucksenkung, die nach alleiniger Anwendung von Sildenafil an gesunden Probanden beobachtet wurde.
In drei spezifischen Arzneimittelinteraktionsstudien wurden der Alphablocker Doxazosin (4 mg und 8 mg) und Sildenafil (25 mg, 50 mg oder 100 mg) gemeinsam an Patienten mit benigner Prostatahyperplasie (BPH) angewendet, die eine Therapie mit Doxazosin in stabiler Dosis erhielten. Bei diesen Studienpopulationen zeigten sich mittlere zusätzliche Senkungen des systolischen und diastolischen Blutdrucks im Liegen um 7/7 mmHg, 9/5 mmHg bzw. 8/4 mmHg und mittlere zusätzliche Senkungen des systolischen und diastolischen Blutdrucks im Stehen um 6/6 mmHg, 11/4 mmHg bzw. 4/5 mmHg. Bei gleichzeitiger Anwendung von Sildenafil und Doxazosin an Patienten mit stabil eingestellter Doxazosin-Dosis gab es gelegentlich Berichte über eine symptomatische orthostatische Hypotonie. Gemeldet wurden dabei Schwindel und Benommenheit, jedoch keine Synkope. Eine gleichzeitige Anwendung von Sildenafil bei Patienten mit Alphablocker-Therapie kann bei empfindlichen Personen zu orthostatischer Hypotonie führen (siehe Abschnitt 4.4).
Sildenafil (100-mg-Einzeldosis) hatte keinen Einfluss auf die Steady-State-Pharmakokinetik des HIV-Protease-Hemmstoffs Saquinavir, der ein CYP3A4-Substrat/Hemmer ist.
Entsprechend seiner pharmakologischen Wirkung auf den Stickstoffmonoxid-cGMP-Stoffwechsel (siehe Abschnitt 5.1) konnte gezeigt werden, dass Sildenafil den blutdrucksenkenden Effekt von Nitraten verstärkt. Daher ist die gleichzeitige Gabe mit Stickstoffmonoxid-Donatoren oder jeglichen Nitraten kontraindiziert (siehe Abschnitt 4.3).
Riociguat
Präklinische Studien zeigten einen additiven Effekt auf die Senkung des systemischen Blutdrucks, wenn PDE5-Inhibitoren mit Riociguat kombiniert wurden. In klinischen Studien zeigte sich, dass Riociguat den hypotensiven Effekt von PDE5-Hemmern verstärkt. Es gab keinen Hinweis auf einen positiven klinischen Effekt dieser Kombination in der untersuchten Studienpopulation. Die gleichzeitige Verwendung von Riociguat zusammen mit PDE5-Hemmern, inklusive Sildenafil, ist kontraindiziert (siehe Abschnitt 4.3).
Sildenafil hatte keinen klinisch relevanten Einfluss auf die Plasmaspiegel von oralen Kontrazeptiva (Ethinylestradiol 30 µg und Levonorgestrel 150 µg).
Die zusätzliche Gabe einer Einzeldosis Sildenafil zu Sacubitril/Valsartan im Steady-State bei Patienten mit Hypertonie war mit einer signifikant stärkeren Blutdrucksenkung verbunden als die Gabe von Sacubitril/Valsartan allein. Daher ist Vorsicht geboten, wenn eine Behandlung mit Sildenafil bei Patienten begonnen wird, die mit Sacubitril/Valsartan behandelt werden.
Kinder und Jugendliche
Interaktionsstudien wurden nur bei Erwachsenen durchgeführt.
Frauen im gebärfähigen Alter und Kontrazeption bei Männern und Frauen
Wegen fehlender Daten zu den Auswirkungen von Revatio bei Schwangeren wird Revatio bei Frauen im gebärfähigen Alter nur dann empfohlen, wenn gleichzeitig ein wirksamer Empfängnisschutz angewandt wird.
Schwangerschaft
Es liegen keine Daten für die Anwendung von Sildenafil bei schwangeren Frauen vor. Tierstudien zeigen keine direkt oder indirekt schädlichen Wirkungen in Bezug auf Schwangerschaft und embryonale/fetale Entwicklung. Studien an Tieren zeigten eine Toxizität hinsichtlich der postnatalen Entwicklung (siehe Abschnitt 5.3).
Aufgrund fehlender Daten sollte Revatio bei schwangeren Frauen nicht angewendet werden, es sei denn, eine Anwendung ist dringend erforderlich.
Stillzeit
Es liegen keine geeigneten und gut kontrollierten Studien an stillenden Frauen vor. Daten von einer stillenden Frau weisen darauf hin, dass Sildenafil und sein aktiver Metabolit N-Desmethylsildenafil in sehr geringen Konzentrationen in die Muttermilch ausgeschieden werden. Es liegen keine klinischen Daten dazu vor, ob Sildenafil nachteilige Auswirkungen auf gestillte Neugeborene/Kinder hat. Die aufgenommenen Mengen lassen jedoch keine nachteiligen Auswirkungen erwarten. Verschreibende Ärzte sollten den klinischen Bedarf der Mutter für eine Anwendung von Sildenafil und mögliche nachteilige Auswirkungen auf den gestillten Säugling sorgfältig gegeneinander abwägen.
Fertilität
Auf der Basis von Standarduntersuchungen zur Fertilität zeigten die präklinischen Daten keine besonderen Risiken für den Menschen (siehe Abschnitt 5.3).
Revatio hat geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Da in klinischen Studien mit Sildenafil über Schwindel und Sehstörungen berichtet wurde, sollen die Patienten darauf achten, wie sie auf die Einnahme von Revatio reagieren, bevor sie ein Fahrzeug lenken oder Maschinen bedienen.
Zusammenfassung des Nebenwirkungsprofils
In der placebokontrollierten Zulassungsstudie mit Revatio bei pulmonaler arterieller Hypertonie wurden insgesamt 207 Patienten auf Revatio in einer Tagesdosis von 20 mg, 40 mg oder 80 mg dreimal täglich randomisiert und 70 Patienten auf Placebo. Die Behandlungsdauer betrug 12 Wochen. Bei den mit 20 mg, 40 mg oder 80 mg Sildenafil dreimal täglich behandelten Patienten betrug die Gesamtabbruchrate 2,9 %, 3,0 % oder 8,5 % im Vergleich zu 2,9 % unter Placebo. Von den 277 Patienten, die in der Zulassungsstudie behandelt wurden, wurden 259 Patienten in eine Langzeit-Fortsetzungsstudie aufgenommen, in der Dosen von bis zu 80 mg dreimal täglich (das 4-Fache der empfohlenen Dosis von 20 mg dreimal täglich) gegeben wurden. Nach 3 Jahren erhielten noch 87 % der 183 Patienten unter Studienmedikation 80 mg Revatio dreimal täglich.
In einer placebokontrollierten Studie mit Revatio als Begleitmedikation zu intravenös verabreichtem Epoprostenol bei pulmonaler arterieller Hypertonie wurden insgesamt 134 Patienten mit Epoprostenol und Revatio (mit einer fixen Dosissteigerung von anfangs 20 mg, dann 40 mg und schließlich 80 mg jeweils dreimal täglich, entsprechend der Verträglichkeit) sowie 131 Patienten mit Epoprostenol und Placebo behandelt. Die Behandlungsdauer betrug 16 Wochen. Die Häufigkeit von Therapieabbrüchen aufgrund unerwünschter Arzneimittelwirkungen lag insgesamt unter Sildenafil/Epoprostenol bei 5,2 %, im Vergleich zu 10,7 % unter Placebo/Epoprostenol. Zu den bis dahin nicht berichteten Nebenwirkungen, die in der Sildenafil/Epoprostenol-Gruppe häufiger auftraten als mit Placebo/Epoprostenol, zählten: okulare Hyperämie, verschwommenes Sehen, Nasenschleimhautschwellung, nächtliche Schweißausbrüche, Rückenschmerzen und Mundtrockenheit. Bekannte Nebenwirkungen wie Kopfschmerzen, Erytheme, Gliederschmerzen und Ödeme wurden häufiger bei mit Sildenafil/Epoprostenol behandelten als bei mit Placebo/Epoprostenol behandelten Patienten beobachtet. Von den Patienten, die diese initiale Studie abschlossen, wurden 242 Patienten in eine Langzeit-Fortsetzungsstudie aufgenommen. Dabei wurden Dosen bis zu 80 mg dreimal täglich gegeben, und nach 3 Jahren erhielten noch 68 % der 133 Patienten unter Studienmedikation 80 mg Revatio dreimal täglich.
In den beiden placebokontrollierten Studien waren die Nebenwirkungen im Allgemeinen leichter bis mäßiger Art. Die am häufigsten beschriebenen Nebenwirkungen (Häufigkeit: 10 % oder größer) mit Revatio im Vergleich zu Placebo waren Kopfschmerzen, Flush, Dyspepsie, Durchfall und Gliederschmerzen.
In einer Studie zur Bewertung der Auswirkungen verschiedener Sildenafil-Dosierungen entsprachen die Sicherheitsdaten für Sildenafil 20 mg dreimal täglich (empfohlene Dosis) und für Sildenafil 80 mg dreimal täglich (das 4-Fache der empfohlenen Dosis) dem Sicherheitsprofil von Sildenafil in früheren PAH-Studien an Erwachsenen.
Tabellarische Auflistung der Nebenwirkungen
Tabelle 1 zeigt die Nebenwirkungen, die bei mindestens 1 % der mit Revatio behandelten Patienten und häufiger (Unterschied > 1 %) als unter Placebo auftraten (Datenbasis ist die Zulassungsstudie zu Revatio bzw. eine gemeinsame Auswertung der beiden placebokontrollierten Studien zu PAH mit Dosierungen von 20, 40 oder 80 mg Sildenafil dreimal täglich). Die Nebenwirkungen sind nach Organsystem und Häufigkeit gegliedert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1.000, < 1/100) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Die Meldungen nach Markteinführung sind kursiv angegeben.
Tabelle 1: Nebenwirkungen aus placebokontrollierten Studien zu Sildenafil bei PAH und aus Meldungen nach Markteinführung bei Erwachsenen
Systemorganklassen gemäß MedDRA (V. 14.0) | Nebenwirkung |
Infektionen und parasitäre Erkrankungen | |
Häufig | Cellulitis, Grippe, Bronchitis, Sinusitis, Rhinitis, Gastroenteritis |
Erkrankungen des Blutes und des Lymphsystems | |
Häufig | Anämie |
Stoffwechsel- und Ernährungsstörungen | |
Häufig | Flüssigkeitsretention |
Psychiatrische Erkrankungen | |
Häufig | Schlaflosigkeit, Angst |
Erkrankungen des Nervensystems | |
Sehr häufig | Kopfschmerzen |
Häufig | Migräne, Tremor, Parästhesie, Brennen, Hypästhesie |
Augenerkrankungen | |
Häufig | Retinablutungen, Sehstörungen, verschwommenes Sehen, Photophobie, Chromopsie, Zyanopsie, Augenreizungen, okuläre Hyperämie |
Gelegentlich | verminderte Sehschärfe, Doppeltsehen, Fremdkörpergefühl im Auge |
Nicht bekannt | nicht arteriitische anteriore ischämische Optikusneuropathie (NAION)*, Verschluss von Netzhautgefäßen*, Gesichtsfelddefekte* |
Erkrankungen des Ohrs und des Labyrinths | |
Häufig | Vertigo |
Nicht bekannt | plötzlicher Hörverlust |
Gefäßerkrankungen | |
Sehr häufig | Flush |
Nicht bekannt | Hypotonie |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Häufig | Nasenbluten, Husten, Nasenschleimhautschwellung |
Erkrankungen des Gastrointestinaltrakts | |
Sehr häufig | Durchfall, Dyspepsie |
Häufig | Gastritis, gastroösophagealer Reflux, Hämorrhoiden, abdominelles Spannungsgefühl, Mundtrockenheit |
Erkrankungen der Haut und des Unterhautzellgewebes | |
Häufig | Alopezie, Erythem, nächtliche Schweißausbrüche |
Nicht bekannt | Ausschlag |
Skelettmuskulatur- und Bindegewebserkrankungen | |
Sehr häufig | Gliederschmerzen |
Häufig | Myalgie, Rückenschmerzen |
Erkrankungen der Nieren und Harnwege | |
Gelegentlich | Hämaturie |
Erkrankungen der Geschlechtsorgane und der Brustdrüse | |
Gelegentlich | Penisblutung, Hämatospermie, Gynäkomastie |
Nicht bekannt | Priapismus, vermehrte Erektionen |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Häufig | Fieber |
*Diese unerwünschten Ereignisse wurden bei Patienten, die PDE5-Hemmer zur Behandlung der erektilen Dysfunktion einnahmen, berichtet.
Kinder und Jugendliche
In der placebokontrollierten Revatio-Studie bei Patienten mit pulmonaler arterieller Hypertonie im Alter von 1 bis 17 Jahren wurden insgesamt 174 Patienten dreimal täglich mit niedrigen Dosen von Revatio (10 mg bei Patienten > 20 kg; kein Patient ≤ 20 kg erhielt diese niedrige Dosis), mittleren Dosen (10 mg bei Patienten ≥ 8 bis 20 kg; 20 mg bei Patienten ≥ 20 bis 45 kg; 40 mg bei Patienten > 45 kg) oder hohen Dosen (20 mg bei Patienten ≥ 8 bis 20 kg; 40 mg bei Patienten ≥ 20 bis 45 kg; 80 mg bei Patienten > 45 kg) behandelt und 60 Patienten erhielten Placebo.
Das in der Studie bei Kindern beobachtete Nebenwirkungsprofil entsprach im Allgemeinen dem bei Erwachsenen (siehe oben stehende Tabelle). Die häufigsten Nebenwirkungen (mit einer Häufigkeit von ≥ 1 %), die bei mit Revatio behandelten Patienten (alle Dosierungen) auftraten, mit einer Häufigkeit von > 1 % gegenüber der Placebo-Gruppe, waren Fieber, Infektionen der oberen Atemwege (je 11,5 %), Erbrechen (10,9 %), vermehrte Erektionen (einschließlich spontaner Erektion des Penis bei männlichen Individuen) (9,0 %), Übelkeit, Bronchitis (je 4,6 %), Pharyngitis (4,0 %), Rhinorrhö (3,4 %) und Pneumonie, Rhinitis (je 2,9 %).
Von den 234 pädiatrischen Patienten, die in der placebokontrollierten Kurzzeit-Studie behandelt wurden, haben 220 Patienten an der Langzeit-Anschlussstudie teilgenommen. Teilnehmer, die eine aktive Sildenafil-Therapie erhalten hatten, haben die gleichen Dosierungsschemata fortgeführt, während die Teilnehmer aus der Placebo-Gruppe der Kurzzeit-Studie randomisiert einer Sildenafil-Behandlung zugeordnet wurden.
Die häufigsten Nebenwirkungen, die während der gesamten Dauer der Kurzzeit- und Langzeit-Studie berichtet wurden, waren im Allgemeinen den Nebenwirkungen ähnlich, die in der Kurzzeit-Studie beobachtet wurden. Nebenwirkungen, die bei > 10 % der 229 mit Sildenafil behandelten Patienten (kombinierte Dosisgruppe, einschließlich 9 Patienten, die die Langzeit-Studie nicht fortgesetzt haben) auftraten, waren Infektion der oberen Atemwege (31 %), Kopfschmerz (26 %), Erbrechen (22 %), Bronchitis (20 %), Pharyngitis (18 %), Fieber (17 %), Diarrhö (15 %) und Grippe, Epistaxis (jeweils 12 %). Der Schweregrad der meisten dieser Nebenwirkungen wurde als leicht bis mäßig eingestuft.
Bei 94 (41 %) der 229 Patienten, die Sildenafil erhielten, wurden schwerwiegende Nebenwirkungen berichtet. Von diesen 94 Patienten, die eine schwerwiegende Nebenwirkung gemeldet haben, waren 14/55 (25,5 %) in der Gruppe mit der geringen Sildenafil-Dosierung, 35/74 (47,3 %) in der Gruppe mit der mittleren Sildenafil-Dosierung und 45/100 (45 %) in der Gruppe mit der hohen Sildenafil-Dosierung. Die häufigsten schwerwiegenden Nebenwirkungen, die mit einer Häufigkeit von ≥ 1 % bei Sidenafil-Patienten (kombinierte Dosierungen) auftraten, waren Pneumonie (7,4 %), Herzversagen, pulmonare Hypertonie (je 5,2 %), Entzündungen der oberen Atemwege (3,1 %), Versagen der rechten Herzkammer, Gastroenteritis (je 2,6 %), Synkope, Bronchitis, Bronchopneumonie, pulmonale arterielle Hypertonie (je 2,2 %), Brustschmerzen, Karies (je 1,7 %) und kardiogener Schock, virale Gastroenteritis, Harnwegsinfektionen (je 1,3 %).
Die folgenden schwerwiegenden Nebenwirkungen wurden als behandlungsbedingt bewertet, Enterokolitis, Konvulsion, Hypersensitivität, Stridor, Hypoxie, neurosensorische Taubheit und ventrikuläre Arrhythmie.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das aufgeführte nationale Meldesystem anzuzeigen.
Deutschland |
In Studien erhielten gesunde Probanden Einzeldosen bis zu 800 mg. Die hierbei beobachteten Nebenwirkungen waren ähnlich wie die bei niedrigeren Dosen, lediglich Inzidenz und Schweregrad waren erhöht. Bei Einzeldosen von 200 mg war die Inzidenz der Nebenwirkungen (Kopfschmerz, Flush, Schwindel, Dyspepsie, Verstopfung der Nase und Sehstörungen) erhöht.
In Fällen einer Überdosierung sollten je nach Bedarf die üblichen unterstützenden Maßnahmen eingeleitet werden. Da Sildenafil in hohem Maße an Plasmaproteine gebunden ist und renal nicht eliminiert wird, ist durch eine Dialyse keine Beschleunigung der Clearance zu erwarten.
Pharmakotherapeutische Gruppe: Urologika, Mittel bei erektiler Dysfunktion
ATC-Code: G04BE03
Wirkmechanismus
Sildenafil ist ein wirksamer und selektiver Hemmstoff der für zyklisches Guanosinmonophosphat (cGMP) spezifischen Phosphodiesterase Typ 5 (PDE5), dem Enzym, das für den Abbau von cGMP verantwortlich ist. Abgesehen von seinem Vorliegen im Corpus cavernosum des Penis findet sich PDE5 auch in den Lungengefäßen. Sildenafil erhöht somit cGMP innerhalb der glatten Muskelzellen der Lungengefäße und führt so zu einer Entspannung. Bei Patienten mit PAH kann dies zu einer selektiven Vasodilatation im pulmonalen Gefäßsystem und – in geringerem Ausmaß – zu einer Vasodilatation im systemischen Kreislauf führen.
Pharmakodynamische Wirkungen
In-vitro-Studien zeigten, dass Sildenafil für PDE5 selektiv ist. Es wirkt stärker auf PDE5 als auf andere bekannte Phosphodiesterasen. Die Selektivität von Sildenafil für PDE5 ist um das 10-Fache höher als für PDE6, die am Phototransduktionsprozess in der Retina beteiligt ist. Es zeigte sich eine 80‑fach höhere Selektivität für PDE5 als für PDE1 und eine um mehr als das 700-Fache höhere Selektivität für PDE5 als für PDE2, 3, 4, 7, 8, 9, 10 und 11. Insbesondere hat Sildenafil eine mehr als 4000‑fach höhere Selektivität für PDE5 im Vergleich zu PDE3, dem an der Steuerung der kardialen Kontraktilität beteiligten cAMP-spezifischen Phosphodiesterase-Isoenzym.
Sildenafil bewirkt eine geringe und vorübergehende Reduktion des Blutdrucks, die in den meisten Fällen keine klinisch relevanten Effekte zur Folge hat.
Nach Langzeitapplikation von 80 mg dreimal täglich an Patienten mit systemischer Hypertonie war die mittlere Veränderung des systolischen und des diastolischen Blutdrucks eine Senkung von 9,4 mmHg bzw. 9,1 mmHg gegenüber dem Ausgangswert.
Nach Langzeitapplikation von 80 mg dreimal täglich an Patienten mit PAH wurden geringere Effekte auf die Blutdrucksenkung beobachtet (eine Senkung von jeweils 2 mmHg systolisch und diastolisch).
Bei der empfohlenen Dosierung von dreimal täglich 20 mg wurde keine Senkung des systolischen oder diastolischen Blutdrucks beobachtet. Die einmalige orale Gabe von bis zu 100 mg Sildenafil ergab bei gesunden Freiwilligen keine Auswirkung auf das EKG. In der Langzeitanwendung von dreimal täglich 80 mg Sildenafil bei Patienten mit PAH wurden keine signifikanten Veränderungen des EKG beobachtet.
In einer Studie zu den hämodynamischen Effekten einer oralen Einmalgabe von 100 mg Sildenafil bei 14 Patienten mit schwerer (> 70%ige Stenose mindestens einer Koronararterie) koronarer Herzkrankheit (KHK) nahmen der mittlere systolische und der mittlere diastolische Blutdruck in Ruhe im Vergleich zum Ausgangswert um 7 % bzw. 6 % ab. Der mittlere pulmonale systolische Blutdruck nahm um 9 % ab. Sildenafil beeinflusste weder das Herzminutenvolumen, noch beeinträchtigte es die Durchblutung in den stenosierten Koronararterien.
Leichte und vorübergehende Veränderungen des Farbsehens (Blau/Grün) wurden bei einigen Studienteilnehmern durch den Farnsworth-Munsell-100-Farben-Test 1 Stunde nach Einnahme von 100 mg beobachtet, 2 Stunden nach Einnahme waren diese Veränderungen nicht mehr nachweisbar. Der vermutete Mechanismus für diese Veränderung des Farbsehens bezieht sich auf die Hemmung der PDE6, die bei dem Phototransduktionsprozess der Retina eine Rolle spielt. Sildenafil übt keinen Einfluss auf die Sehschärfe oder das Kontrastsehen aus. In einer kleinen, placebokontrollierten Untersuchung bei 9 Patienten mit dokumentierter altersbedingter Makuladegeneration im Frühstadium zeigte Sildenafil als 100-mg-Einmaldosis in den durchgeführten Sehtests (Sehschärfe, Amsler-Gitter, Lichtertest, Humphrey-Perimeter und Photostress-Test) keine signifikanten Veränderungen.
Klinische Wirksamkeit und Sicherheit
Wirksamkeit bei erwachsenen Patienten mit PAH
Es wurde eine randomisierte, doppelblinde, placebokontrollierte Studie bei 278 Patienten mit primärer PAH, PAH in Verbindung mit einer Bindegewebskrankheit und PAH nach chirurgischer Korrektur eines angeborenen Herzfehlers durchgeführt. Die Patienten wurden für eine von 4 Behandlungsgruppen randomisiert: Placebo, Sildenafil 20 mg, Sildenafil 40 mg oder Sildenafil 80 mg, je dreimal täglich. Von den 278 randomisierten Patienten erhielten 277 zumindest 1 Dosis der Studienmedikation. Die Studienpopulation umfasste 68 (25 %) Männer und 209 (75 %) Frauen mit einem mittleren Alter von 49 Jahren (Altersbereich: 18 bis 81 Jahre) und einer 6-Minuten-Gehstrecke zwischen 100 und 450 Meter (Mittelwert: 344 Meter) bei Studienbeginn. 175 Patienten (63 %) hatten eine Diagnose mit primärer pulmonaler Hypertonie, 84 (30 %) eine Diagnose mit PAH in Verbindung mit einer Bindegewebskrankheit und 18 (7 %) eine Diagnose mit PAH nach einer chirurgischen Korrektur eines angeborenen Herzfehlers. Die meisten Patienten gehörten bei Studienbeginn in die WHO-Funktionsklasse II (107/277; 39 %) oder III (160/277; 58 %) und wiesen eine durchschnittliche 6-Minuten-Gehstrecke von 378 Meter bzw. 326 Meter auf, weniger Patienten in die Funktionsklasse I (1/277; 0,4 %) oder IV (9/277; 3 %). Patienten mit einer linksventrikulären Auswurffraktion < 45 % oder mit linksventrikulärer Verkürzungsfraktion < 0,2 % waren von einer Teilnahme ausgeschlossen.
Sildenafil (oder Placebo) wurde zusätzlich zur bestehenden Therapie der Patienten verabreicht, die eine Kombination von Antikoagulanzien, Digoxin, Calciumantagonisten, Diuretika oder Sauerstoff umfassen konnte. Die Anwendung von Prostacyclin, Prostacyclinanaloga oder Endothelinantagonisten als Zusatzbehandlung war ebenso wenig gestattet wie eine Argininsupplementation. Patienten, die zuvor auf eine Therapie mit Bosentan nicht angesprochen hatten, waren von einer Teilnahme an der Studie ausgeschlossen.
Der primäre Endpunkt für die Wirksamkeit war die Veränderung der 6-Minuten-Gehstrecke in Woche 12 gegenüber dem Ausgangswert. Für alle drei Sildenafil-Dosis-Gruppen zeigte sich im Vergleich zu den Patienten mit Placebo eine statistisch signifikante Erhöhung der 6-Minuten-Gehstrecke. Die relative Erhöhung der 6-Minuten-Gehstrecke gegenüber Placebo betrug 45 Meter (p < 0,0001), 46 Meter (p < 0,0001) bzw. 50 Meter (p < 0,001) für Sildenafil 20 mg, 40 mg bzw. 80 mg dreimal täglich. Es gab keinen signifikanten Unterschied in der Wirkung zwischen den einzelnen Dosen von Sildenafil. Bei Patienten mit einem Ausgangwert der 6-Minuten-Gehstrecke unter 325 Meter wurde eine verbesserte Wirksamkeit bei den höheren Dosen beobachtet (die Verbesserung gegenüber Placebo betrug 58 Meter, 65 Meter bzw. 87 Meter für 20 mg, 40 mg bzw. 80 mg dreimal täglich).
Unter Berücksichtigung der WHO-Funktionsklassen konnte in der 20-mg-Dosis-Gruppe eine statistisch signifikante Erhöhung der 6-Minuten-Gehstrecke beobachtet werden: Für die Funktionsklassen II und III wurden gegenüber Placebo Erhöhungen um 49 Meter (p = 0,0007) und 45 Meter (p = 0,0031) gemessen.
Die Verbesserung der 6-Minuten-Gehstrecke war bereits nach 4 Wochen Behandlung eindeutig feststellbar und konnte auch über 8 und 12 Wochen aufrechterhalten werden. Die Therapieeffekte waren bei den verschiedenen Subgruppen vergleichbar, wobei die Subgruppen nach der Ätiologie (primäre PAH und pulmonale Hypertonie in Verbindung mit einer Bindegewebskrankheit), den verschiedenen WHO-Funktionsklassen, Geschlecht, Rasse, den geographischen Regionen, dem mittleren Pulmonalarteriendruck und dem pulmonalen Gefäßwiderstand definiert waren.
Bei allen Dosierungen von Sildenafil zeigten die Patienten eine statistisch signifikante Reduktion des mittleren Pulmonalarteriendrucks (mPAP) und des pulmonalen Gefäßwiderstands (PVR) im Vergleich zu denen mit Placebo. Die placebokorrigierten Behandlungseffekte auf den mPAP betrugen -2,7 mmHg (p = 0,04), -3,0 mmHg (p = 0,01) bzw. -5,1 mmHg (p < 0,0001) für dreimal täglich 20 mg, 40 mg bzw. 80 mg Sildenafil. Die gegenüber Placebo relativen Behandlungseffekte auf den PVR betrugen -178 dyn.sec/cm5 (p = 0,0051), -195 dyn.sec/cm5 (p = 0,0017) bzw. -320 dyn.sec/cm5 (p < 0,0001) für dreimal täglich 20 mg, 40 mg bzw. 80 mg Sildenafil. Nach 12 Wochen mit dreimal täglich 20 mg, 40 mg bzw. 80 mg Sildenafil war die prozentuale Senkung des PVR proportional größer (11,2 %, 12,9 % bzw. 23,3 %) als die Reduktion für den systemischen Gefäßwiderstand (7,2 %, 5,9 % bzw. 14,4 %). Der Einfluss von Sildenafil auf die Mortalität ist nicht bekannt.
Bei allen Sildenafil-Dosierungen ergab sich in Woche 12 bei einem größeren Prozentsatz der Patienten (nämlich bei 28 %, 36 % bzw. 42 % der Personen unter dreimal täglich 20 mg, 40 mg bzw. 80 mg) eine Verbesserung um mindestens 1 WHO-Funktionsklasse im Vergleich zu 7 % unter Placebo. Die jeweilige Odds-Ratio betrug 2,92 (p = 0,0087), 4,32 (p = 0,0004) bzw. 5,75 (p < 0,0001).
Langzeit-Überlebensdaten bei nicht vorbehandelten Patienten
Die Patienten der Zulassungsstudie konnten als Fortsetzung an einer offenen Langzeitstudie teilnehmen. Nach 3 Jahren erhielten 87 % der Patienten eine Dosierung von dreimal täglich 80 mg. In der Zulassungsstudie wurden insgesamt 207 Patienten mit Revatio behandelt, und ihre Langzeit-Überlebensrate wurde über mindestens 3 Jahre verfolgt. In dieser Patientenpopulation betrug die Kaplan-Meier-Schätzung für die 1-Jahres-, 2-Jahres- und 3-Jahres-Überlebensrate 96 %, 91 % und 82 %. Bei Patienten mit einer WHO-Funktionsklasse II zu Studienbeginn betrug die Überlebensrate nach 1, 2 und 3 Jahren 99 %, 91 % und 84 % und bei Patienten mit einer WHO-Funktionsklasse III zu Studienbeginn 94 %, 90 % und 81 %.
Wirksamkeit bei erwachsenen Patienten mit PAH (bei Kombination mit Epoprostenol)
Es wurde eine randomisierte, placebokontrollierte Doppelblindstudie mit 267 PAH-Patienten durchgeführt, die mit intravenös verabreichtem Epoprostenol eingestellt waren. Die Studienpopulation bestand sowohl aus Patienten mit primärer pulmonaler arterieller Hypertonie (212/267, 79 %) als auch aus Patienten mit PAH in Verbindung mit einer Bindegewebskrankheit (55/267, 21 %). Die meisten Patienten entfielen zu Studienbeginn auf die WHO-Funktionsklassen II (68/267, 26 %) und III (175/267, 66 %), weniger Patienten waren der Funktionsklasse I (3/267, 1 %) oder IV (16/267, 6 %) zugeordnet, und bei einigen Patienten (5/267, 2 %) war die WHO-Funktionsklasse unbekannt. Die Patienten wurden in zwei Gruppen randomisiert: intravenös verabreichtes Epoprostenol plus Placebo oder plus Sildenafil (bei einer fixen Dosissteigerung von anfangs 20 mg, dann 40 mg und schließlich 80 mg jeweils dreimal täglich, entsprechend der Verträglichkeit).
Der primäre Endpunkt für die Wirksamkeit war die Veränderung der 6-Minuten-Gehstrecke in Woche 16 gegenüber dem Ausgangswert. Mit Sildenafil zeigte sich im Vergleich zu Placebo eine statistisch signifikante Erhöhung der 6-Minuten-Gehstrecke. Die mittlere placebokorrigierte Verlängerung der Gehstrecke unter Sildenafil betrug 26 Meter (95%-KI: 10,8 bis 41,2; p = 0,0009). Bei Patienten mit einer Gehstrecke von ≥ 325 Metern zu Studienbeginn war der Behandlungseffekt 38,4 Meter zugunsten von Sildenafil; bei Patienten mit einer Gehstrecke von < 325 Metern zu Studienbeginn war der Behandlungseffekt 2,3 Meter zugunsten von Placebo. Bei Patienten mit primärer PAH war der Behandlungseffekt 31,1 Meter im Vergleich zu 7,7 Metern bei Patienten mit PAH in Verbindung mit einer Bindegewebskrankheit. Aufgrund der geringen Fallzahlen in den einzelnen randomisierten Untergruppen könnten diese Unterschiede auch zufällig sein.
Im Vergleich zu Placebo wurde bei den Patienten unter Sildenafil eine statistisch signifikante Senkung des mittleren Pulmonalarteriendrucks (mPAP) erreicht. Dabei war Sildenafil überlegen mit einer mittleren placebokorrigierten Senkung von -3,9 mmHg (95 %-KI: -5,7 bis -2,1; p = 0,00003). Ein sekundärer Endpunkt war die Zeit bis zum Eintreten einer klinischen Verschlechterung, die als die Zeitspanne von der Randomisierung bis zum ersten Auftreten eines die Erkrankung verschlechternden Ereignisses definiert war (Tod, Lungentransplantation, Beginn einer Bosentan-Therapie oder eine klinische Verschlechterung, die eine Veränderung der Epoprostenol-Therapie notwendig machte). Im Vergleich zu Placebo führte die Behandlung mit Sildenafil zu einer signifikanten Verlängerung der Zeit bis zum Eintreten einer klinischen Verschlechterung der PAH (p = 0,0074). In der Placebo-Gruppe kam es bei 23 Personen zum Auftreten von die Erkrankung verschlechternden Ereignissen (17,6 %) im Vergleich zu 8 Personen in der Sildenafil-Gruppe (6,0 %).
Langzeit-Überlebensdaten aus der Studie bei bestehender Epoprostenol-Therapie
Die an der Studie mit bestehender Epoprostenol-Therapie eingeschlossenen Patienten konnten als Fortsetzung an einer offenen Langzeitstudie teilnehmen. Nach 3 Jahren erhielten 68 % der Patienten eine Dosierung von dreimal täglich 80 mg. Zu Studienbeginn wurden insgesamt 134 Patienten mit Revatio behandelt, und ihre Langzeit-Überlebensdaten wurden über mindestens 3 Jahre verfolgt. In dieser Patientenpopulation betrugen die Kaplan-Meier-Schätzwerte der 1-, 2- und 3-Jahres-Überlebensrate 92 %, 81 % und 74 %.
Wirksamkeit und Sicherheit bei erwachsenen Patienten mit PAH (bei gleichzeitiger Anwendung von Bosentan)
Es wurde eine randomisierte, placebokontrollierte Doppelblindstudie mit 103 klinisch stabilen PAH-Patienten (WHO FK II und III) durchgeführt, die seit mindestens 3 Monaten eine Bosentan-Therapie erhalten hatten. Darunter waren Patienten mit primärer PAH und solche mit PAH in Verbindung mit einer Bindegewebskrankheit. Die Patienten wurden randomisiert einer Behandlung mit Placebo oder Sildenafil (20 mg dreimal täglich) in Kombination mit Bosentan (62,5 bis 125 mg zweimal täglich) zugeordnet. Der primäre Endpunkt für die Wirksamkeit war eine Veränderung der 6-Minuten-Gehstrecke in Woche 12 gegenüber dem Ausgangswert. Die Ergebnisse zeigen, dass kein signifikanter Unterschied in der mittleren Veränderung der 6-Minuten-Gehstrecke gegenüber dem Ausgangswert zwischen Sildenafil 20 mg dreimal täglich (13,62 Meter [KI 95 %: -3,89 bis +31,12]) und Placebo (14,08 Meter [KI 95 %: -1,78 bis +29,95]) zu beobachten ist.
Unterschiede bei der 6-Minuten-Gehstrecke wurden beobachtet zwischen Patienten mit primärer PAH und PAH in Verbindung mit einer Bindegewebskrankheit. Bei Patienten mit primärer PAH (67 Teilnehmer) betrug die mittlere Veränderung gegenüber dem Ausgangswert 26,39 Meter (KI 95 %: 10,70 bis 42,08) in der Sildenafil-Gruppe und 11,84 Meter (KI 95 %: -8,83 bis 32,52) in der Placebo-Gruppe. Bei Patienten mit PAH in Verbindung mit einer Bindegewebskrankheit (36 Teilnehmer) betrug die mittlere Veränderung gegenüber dem Ausgangswert jedoch -18,32 Meter (KI 95 %: -65,66 bis 29,02) in der Sildenafil-Gruppe und 17,50 Meter (KI 95 %: -9,41 bis 44,41) in der Placebo-Gruppe.
Insgesamt waren die Nebenwirkungen in den beiden Behandlungsgruppen (Sildenafil plus Bosentan gegenüber Bosentan alleine) grundsätzlich ähnlich und entsprachen dem bekannten Sicherheitsprofil von Sildenafil als Monotherapie (siehe Abschnitte 4.4 und 4.5).
Auswirkungen auf die Mortalität bei Erwachsenen mit PAH
Nachdem bei Kindern und Jugendlichen nach der Einnahme einer hohen Dosis Sildenafil dreimal täglich, bezogen auf das Körpergewicht, ein höheres Mortalitätsrisiko beobachtet worden war als bei Kindern und Jugendlichen, die in der Langzeit-Anschlussstudie der pädiatrischen klinischen Studie eine niedrigere Dosis erhielten, wurde eine Studie zur Untersuchung der Auswirkungen verschiedener Dosierungen von Sildenafil auf die Mortalität bei Erwachsenen mit PAH durchgeführt (siehe unten unter „Kinder und Jugendliche – Pulmonale arterielle Hypertonie“ und „Daten der Langzeit-Anschlussstudie“).
Es handelte sich um eine randomisierte, doppelblinde Parallelgruppenstudie an 385 Erwachsenen mit PAH. Die Patienten wurden nach dem Zufallsprinzip im Verhältnis von 1:1:1 einer von drei Dosisgruppen zugewiesen (5 mg dreimal täglich [4-mal niedriger als die empfohlene Dosis], 20 mg dreimal täglich [empfohlene Dosis] und 80 mg dreimal täglich [4-Fache der empfohlenen Dosis]). Insgesamt war die PAH bei den meisten Teilnehmern zuvor noch nicht behandelt worden (83,4 %). Bei den meisten Teilnehmern lag eine idiopathische PAH vor (71,7 %). Die häufigste WHO-Funktionsklasse war die Klasse III (57,7 % der Teilnehmer). Alle drei Behandlungsgruppen waren in Bezug auf die demografischen Merkmale der Teilnehmer wie Vorgeschichte mit PAH-Therapie, Ätiologie der PAH sowie WHO-Funktionsklassen gut ausgewogen.
Die Mortalitätsrate betrug 26,4 % (n = 34) für die Dosis mit 5 mg dreimal täglich, 19,5 % (n = 25) für die Dosis mit 20 mg dreimal täglich und 14,8 % (n = 19) für die Dosis mit 80 mg dreimal täglich.
Kinder und Jugendliche
Pulmonale arterielle Hypertonie
In einer randomisierten, doppelblinden, placebokontrollierten, parallelen Multizenterstudie mit verschiedenen Dosierungen wurden insgesamt 234 Personen im Alter von 1 bis 17 Jahren behandelt. Die Teilnehmer (38 % männlich und 62 % weiblich) hatten ein Körpergewicht ≥ 8 kg und litten zu 33 % an primärer pulmonaler Hypertonie (PPH) oder einer PAH in Verbindung mit angeborenen Herzerkrankungen (systemisch-pulmonale Shunts 37 %, chirurgische Reposition 30 %). 63 der 234 Patienten (27 %) in dieser Studie waren jünger als 7 Jahre (niedrige Sildenafil-Dosis: n = 2; mittlere Sildenafil-Dosis: n = 17; hohe Sildenafil-Dosis: n = 28; Placebo: n = 16), und 171 der 234 Patienten (73 %) waren 7 Jahre oder älter (niedrige Sildenafil-Dosis: n = 40; mittlere Sildenafil-Dosis: n = 38; hohe Sildenafil-Dosis: n = 49; Placebo: n = 44). Die meisten Personen waren als Ausgangwert in der WHO-Funktionsklasse I (75/234, 32 %) oder II (120/234, 51 %). Weniger Patienten waren Funktionsklasse III (35/234, 15 %) oder IV (1/234, 0,4 %). Bei einigen wenigen Patienten (3/234, 1,3 %) war die WHO-Funktionsklasse nicht bekannt.
Die Patienten waren nicht mit einer spezifischen PAH-Therapie vorbehandelt und die Anwendung von Prostacyclin, Prostacyclinanaloga sowie Endothelinrezeptor-Antagonisten waren in der Studie nicht erlaubt, ebenso wenig wie Argininsupplementierung, Nitrate, Alphablocker und starke CYP450-3A4-Hemmer.
Das primäre Ziel der Studie war, die Wirksamkeit von oralem Sildenafil auf die Verbesserung der körperlichen Belastbarkeit bei Kindern in der Dauertherapie über 16 Wochen anhand des Cardiopulmonary-Exercise-Tests (CPET) bei den Teilnehmern, die von ihrer Entwicklung her dazu imstande waren (n = 115) zu prüfen. Die sekundären Endpunkte schlossen u. a. ein hämodynamisches Monitoring, Erfassung der Symptome, die WHO-Funktionsklasse, Veränderung der Begleitmedikation und die Erfassung der Lebensqualität mit ein.
Die Teilnehmer wurden entweder einer der drei Sildenafil-Gruppen zugeteilt (niedrige [10 mg], mittlere [10 bis 40 mg] oder hohe [20 bis 80 mg] Revatio-Dosen dreimal täglich) oder auf die Placebo-Gruppe. Die innerhalb einer Gruppe tatsächlich gegebene Dosis orientierte sich am Körpergewicht (siehe Abschnitt 4.8). Der Anteil der Teilnehmer, die zu Beginn eine unterstützende Therapie (Antikoagulanzien, Digoxin, Calciumkanalblocker, Diuretika und/oder Sauerstoff) erhielten, war in der kombinierten Sildenafil-Gruppe (47,7 %) und in der Placebo-Gruppe (41,7 %) vergleichbar.
Der primäre Endpunkt war die durch CPET in den kombinierten Sildenafil-Gruppen erhobene, placebokorrigierte prozentuale Veränderung des max. VO2 in Woche 16 gegenüber dem Ausgangswert (siehe Tabelle 2). Insgesamt waren 106 von den 234 Teilnehmern (45 %) mittels CPET auswertbar. Es handelte sich hierbei um die Kinder, die 7 Jahre und älter und von ihrer Entwicklung her imstande waren, den Test durchzuführen. Bei den Kindern unter 7 Jahren (kombinierte Sildenafil-Gruppe: n = 47; Placebo-Gruppe: n = 16) konnten nur die sekundären Endpunkte erhoben werden. Die durchschnittlichen Ausgangswerte für die max. Sauerstoffaufnahme waren innerhalb der Sildenafil-Gruppen vergleichbar (17,37 bis 18,03 ml/kg/min) und in der Placebo-Gruppe geringfügig höher (20,02 ml/kg/min). Die Ergebnisse der Gesamtauswertung (kombinierte Dosis-Gruppe vs. Placebo) unterschieden sich nicht signifikant (p = 0,056; siehe Tabelle 2). Zwischen der mittleren Sildenafil-Dosis und Placebo betrug der berechnete Unterschied 11,33 % (95%-KI: 1,72 bis 20,94; siehe Tabelle 2).
Tabelle 2: Placebokorrigierte prozentuale Veränderung des Peak-VO2 gegenüber dem Ausgangswert in den aktiven Behandlungsgruppen.
Behandlungsgruppe | Berechneter Unterschied | 95 %-Konfidenzintervall |
Niedrige Dosis (n = 24) | 3,81 | -6,11 bis 13,73 |
Mittlere Dosis (n = 26) | 11,33 | 1,72 bis 20,94 |
Hohe Dosis (n = 27) | 7,98 | -1,64 bis 17,60 |
Kombinierte Dosen (n = 77) | 7,71 (p = 0,056) | -0,19 bis 15,60 |
Placebo-Gruppe: n = 29
Schätzwerte beruhend auf ANCOVA mit den Kovariablen Ausgangswert der max. VO2, Ätiologie und Gewichtsgruppe
Dosisabhängige Verbesserungen wurden beim pulmonalen Gefäßwiderstands-Index (PVRI) und dem durchschnittlichen pulmonalen arteriellen Druck (mPAP) beobachtet. Mit -18 % (95%-KI: 2 % bis 32 %) und -27 % (95 %-KI: 14 % bis 39 %) gegenüber der Placebo-Gruppe ergab sich in den Sildenafil-Gruppen mit mittlerer und hoher Dosis eine Verringerung des PVRI. Die Gruppe mit der niedrigen Dosis zeigte keine signifikanten Unterschiede gegenüber der Placebo-Gruppe (Unterschied: 2 %). Mit -3,5 mmHg (95%-KI: -8,9 bis 1,9) und -7,3 mmHg (95%-KI: -12,4 bis -2,1) gegenüber dem Ausgangswert ergab sich in den Sildenafil-Gruppen mit mittlerer und hoher Dosis eine Veränderung des mPAP im Vergleich zur Placebo-Gruppe. Die Gruppe mit der niedrigen Dosis zeigte nur kleine Unterschiede gegenüber Placebo (Unterschied: 1,6 mmHg). Alle drei Sildenafil-Gruppen zeigten gegenüber Placebo eine Verbesserung des Herzindex von 10 %, 4 % und 15 % jeweils für die Gruppe mit niedriger, mittlerer und hoher Dosis.
Gegenüber Placebo zeigte sich lediglich bei den Teilnehmern mit der hohen Sildenafil-Dosis eine signifikante Verbesserung der Funktionsklasse. Im Vergleich zu Placebo betrug die Odds-Ratio in den Sildenafil-Gruppen mit niedriger, mittlerer und hoher Dosis 0,6 (95%-KI: 0,18 bis 2,01), 2,25 (95%-KI: 0,75 bis 6,69) und 4,52 (95%-KI: 1,56 bis 13,10).
Daten der Langzeit-Anschlussstudie
Von den 234 pädiatrischen Patienten, die in der placebokontrollierten Kurzzeit-Studie behandelt wurden, haben 220 Patienten an der Langzeit-Anschlussstudie teilgenommen. Dabei wurden Teilnehmer aus der Placebo-Gruppe der Kurzzeit-Studie randomisiert einer Sildenafil-Behandlung zugeordnet; Patienten mit einem Gewicht ≤ 20 kg wurden in die Gruppen mit mittlerer oder hoher Dosis (1:1) aufgenommen, während Patienten mit einem Gewicht > 20 kg in die niedrige, mittlere oder hohe Dosisgruppe (1:1:1) aufgenommen wurden. Von den insgesamt 229 Patienten, die Sildenafil erhielten, waren 55, 74 und 100 Patienten in den Gruppen mit niedriger, mittlerer bzw. hoher Dosis. Die Gesamtbehandlungsdauer während der Kurzzeit- und Langzeit-Studien, beginnend mit der Doppelverblindung für die einzelnen Patienten, lag zwischen 3 und 3.129 Tagen. In der Gruppe mit Sildenafil-Behandlung betrug die mittlere Dauer der Sildenafil-Behandlung 1.696 Tage (darin nicht enthalten die 5 Patienten, die in der doppelblinden Phase Placebo erhielten und nicht in der Langzeit-Anschlussstudie behandelt wurden).
In den Gruppen mit niedriger, mittlerer und hoher Dosis betrugen die Kaplan-Meier-Schätzwerte der 3-Jahres-Überlebensrate für die Patienten mit einem Gewicht > 20 kg bei Studienbeginn 94 %, 93 % und 85 %. In den Gruppen mit mittlerer und hoher Dosis betrugen die Schätzwerte der Überlebensrate für die Patienten mit einem Gewicht ≤ 20 kg bei Studienbeginn 94 % und 93 % (siehe Abschnitte 4.4 und 4.8).
Während der Studiendurchführung wurden insgesamt 42 Todesfälle gemeldet, entweder im Verlauf der Behandlung oder während der Nachbeobachtung des Überlebens. 37 Todesfälle traten auf, bevor das Datenüberwachungskomitees (Data Monitoring Committee) die Entscheidung gefällt hatte, die Dosis der Patienten auf eine niedrigere Dosierung zu reduzieren. Diese Entscheidung basierte auf einem beobachteten Ungleichgewicht der Sterblichkeit mit zunehmenden Sildenafil-Dosen. Von diesen 37 Todesfällen betrug die Anzahl (%) in der Gruppe mit niedriger Sildenafil-Dosis 5/55 (9,1 %), mit mittlerer Sildenafil-Dosis 10/74 (13,5 %) und mit hoher Sildenafil-Dosis 22/100 (22 %). Im Anschluss wurden 5 weitere Todesfälle gemeldet. Die Todesursachen wurden mit der PAH in Verbindung gebracht. Höhere als die empfohlenen Dosen dürfen bei pädiatrischen Patienten mit PAH nicht angewendet werden (siehe Abschnitte 4.2 und 4.4).
1 Jahr nach Beginn der placebokontrollierten Studie wurde die max. VO2 bestimmt. Von den mit Sildenafil behandelten Personen, die von ihrer Entwicklung her imstande waren, den CPET durchzuführen, zeigte sich bei 59/114 Personen (52 %) gegenüber dem Zeitpunkt zu Beginn der Sildenafil-Behandlung keinerlei Verschlechterung der max. VO2. In ähnlicher Weise hatte sich die WHO-Funktionsklasse bei 191 von 229 Personen (83 %), die Sildenafil erhalten hatten, bei der Beurteilung nach 1 Jahr unverändert erhalten oder verbessert.
Persistierende pulmonale Hypertonie des Neugeborenen
Eine randomisierte, doppelblinde, zweiarmige, placebokontrollierte Parallelgruppenstudie wurde bei 59 Neugeborenen mit persistierender pulmonaler Hypertonie des Neugeborenen (PPHN) oder mit hypoxischer Ateminsuffizienz und mit Risiko für eine PPHN mit Oxigenierungsindex (OI) > 15 und < 60 durchgeführt. Das primäre Ziel war die Wirksamkeit und Sicherheit von i. v. Sildenafil zu untersuchen, wenn es zusammen mit inhalativem Stickstoffmonoxid (iNO) im Vergleich zu iNO alleine gegeben wurde.
Die Co-primären Endpunkte waren die Therapieversagensrate, definiert als Notwendigkeit zusätzlicher Therapiemaßnahmen gegen PPHN, Notwendigkeit einer extrakorporalen Membranoxygenierung (ECMO) oder Tod während der Studie, sowie die Zeit mit iNO-Therapie nach Initiierung der i. v. Studienmedikation für Patienten ohne Therapieversagen. Die Unterschiede in der Rate an Therapieversagern zwischen den beiden Behandlungsgruppen war statistisch nicht signifikant (27,6 % bzw. 20,0 % in der iNO- + i. v. Sildenafil-Gruppe bzw. iNO- + Placebo-Gruppe). Für Patienten ohne Therapieversagen war die mittlere Zeit unter iNO-Behandlung nach Initiierung der i. v. Studienmedikation mit etwa 4,1 Tagen in beiden Behandlungsgruppen die gleiche.
Therapiebedingte bzw. schwerwiegende unerwünschte Ereignisse wurden bei 22 (75,9 %) bzw. 7 (24,1 %) Teilnehmern in der iNO- + i. v. Sildenafil-Behandlungsgruppe und bei 19 (63,3 %) bzw. 2 (6,7 %) Teilnehmern in der iNO- + Placebo-Gruppe beobachtet. Die häufigsten therapiebedingten unerwünschten Ereignisse waren Hypotonie (8 [27,6 %] Teilnehmer), Hypokaliämie (7 [24,1 %] Teilnehmer), Anämie und Entzugssymptome (jeweils 4 [13,8 %] Teilnehmer) und Bradykardie (3 [10,3 %] Teilnehmer) in der iNO- + i. v. Sildenafil-Behandlungsgruppe und Pneumothorax (4 [13,3 %] Teilnehmer), Anämie, Ödeme, Hyperbilirubinämie, erhöhtes C-reaktives Protein und Hypotonie (jeweils 3 [10 %] Teilnehmer) in der iNO- + Placebo-Behandlungsgruppe (siehe Abschnitt 4.2).
Resorption
Sildenafil wird rasch resorbiert. Die maximalen Plasmaspiegel werden innerhalb von 30 bis 120 Minuten (Median: 60 Minuten) nach oraler Gabe im nüchternen Zustand erreicht. Die mittlere absolute orale Bioverfügbarkeit beträgt 41 % (Streubreite: 25 bis 63 %). Nach dreimal täglicher oraler Einnahme von Sildenafil nehmen AUC und Cmax dosisproportional über den Dosisbereich von 20 bis 40 mg zu. Nach oralen Dosen von 80 mg dreimal täglich wurde ein höherer als dosisproportionaler Anstieg der Plasmaspiegel von Sildenafil beobachtet. Bei Patienten mit PAH war die orale Bioverfügbarkeit von Sildenafil nach einer Dosis von 80 mg dreimal täglich durchschnittlich 43 % (90%-KI: 27 % bis 60 %) höher als mit den niedrigeren Dosen.
Bei Einnahme von Sildenafil zusammen mit einer Mahlzeit ist die Resorptionsrate reduziert, die tmax verzögert sich im Mittel um 60 Minuten, während die Cmax im Mittel um 29 % verringert ist; allerdings war das Ausmaß der Resorption nicht signifikant beeinträchtigt (AUC verringerte sich um 11 %).
Verteilung
Das mittlere Verteilungsvolumen im Steady State (Vss) von Sildenafil beträgt 105 l, was auf eine Verteilung in die Gewebe hinweist. Nach oraler Gabe einer 20-mg-Dosis dreimal täglich beträgt die mittlere maximale Gesamtplasmakonzentration von Sildenafil ca. 113 ng/ml. Sildenafil und sein wichtigster im Blutkreislauf zirkulierender, N-demethylierter Metabolit sind zu etwa 96 % an Plasmaproteine gebunden. Die Proteinbindung ist unabhängig von der Gesamtkonzentration des Arzneimittels.
Biotransformation
Sildenafil wird überwiegend hepatisch durch die mikrosomalen Isoenzyme CYP3A4 (Hauptweg) und CYP2C9 (Nebenweg) metabolisiert. Der wichtigste zirkulierende Metabolit resultiert aus der N-Demethylierung von Sildenafil. Das Profil der Phosphodiesterase-Selektivität dieses Metaboliten ist ähnlich jenem von Sildenafil, und er zeigt eine In-vitro-Hemmwirkung für PDE5, die rund 50 % derjenigen der Stammsubstanz beträgt. Der N-Demethyl-Metabolit wird weiter verstoffwechselt, die terminale Halbwertszeit beträgt rund 4 Stunden. Bei Patienten mit PAH betragen die Plasmaspiegel des N-Demethyl-Metaboliten nach einer Gabe von 20 mg dreimal täglich etwa 72 % jener von Sildenafil (was einem Beitrag von 36 % zu den pharmakologischen Wirkungen von Sildenafil entspricht). Der weitere Effekt auf die Wirksamkeit ist nicht bekannt.
Elimination
Die gesamte Clearance von Sildenafil beträgt 41 l/h mit einer daraus resultierenden terminalen Halbwertszeit von 3 bis 5 Stunden. Nach oraler oder intravenöser Applikation wird Sildenafil nach Metabolisierung hauptsächlich über die Fäzes (rund 80 % der verabreichten oralen Dosis) und in geringerem Maße renal (rund 13 % der verabreichten oralen Dosis) ausgeschieden.
Pharmakokinetik bei speziellen Patientengruppen
Ältere Patienten
Gesunde ältere Freiwillige (65 Jahre oder älter) zeigten eine herabgesetzte Sildenafil-Clearance, wobei die Plasmaspiegel von Sildenafil und des aktiven N-Demethyl-Metaboliten ungefähr 90 % höher lagen als bei jüngeren gesunden Freiwilligen (18 bis 45 Jahre). Aufgrund der altersabhängigen Unterschiede in der Plasmaproteinbindung lag der entsprechende Anstieg der Plasmaspiegel von freiem Sildenafil bei rund 40 %.
Patienten mit Nierenfunktionsstörungen
Bei Probanden mit leichter bis mäßiger Nierenfunktionsstörung (Kreatinin-Clearance = 30 bis 80 ml/min) war die Pharmakokinetik nach einer oralen Sildenafil-Einzeldosis von 50 mg unverändert. Bei Probanden mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min) war die Clearance von Sildenafil herabgesetzt und resultierte in Erhöhungen von AUC und Cmax um 100 % bzw. 88 % im Vergleich zu Probanden gleichen Alters ohne eingeschränkte Nierenfunktion. Zusätzlich waren die AUC und Cmax des N-Demethyl-Metaboliten bei Patienten mit schwerer Nierenfunktionsstörung im Vergleich zu Probanden mit normaler Nierenfunktion signifikant um 200 % bzw. 79 % erhöht.
Patienten mit Leberfunktionsstörungen
Bei Probanden mit leichter bis mäßiger Leberzirrhose (Child-Pugh-Klassen A und B) war die Clearance von Sildenafil herabgesetzt, was zu Erhöhungen von AUC (85 %) und Cmax (47 %) führte, im Vergleich zu Probanden gleichen Alters ohne eingeschränkte Leberfunktion. Zusätzlich waren AUC und Cmax für den N-Demethyl-Metaboliten bei Probanden mit Leberzirrhose im Vergleich zu Probanden mit normaler Leberfunktion signifikant um 154 % bzw. 87 % erhöht. Die Pharmakokinetik von Sildenafil bei Patienten mit schwerer Leberfunktionsstörung wurde nicht untersucht.
Pharmakokinetik bei verschiedenen Patientengruppen
Bei Patienten mit PAH waren die durchschnittlichen Plasmaspiegel im Steady State über den untersuchten Dosisbereich von 20 bis 80 mg dreimal täglich um 20 bis 50 % höher als bei gesunden Freiwilligen. Es zeigte sich eine Verdoppelung der Cmin im Vergleich zu gesunden Freiwilligen. Diese beiden Befunde lassen eine geringere Clearance und/oder eine höhere orale Bioverfügbarkeit von Sildenafil bei Patienten mit PAH im Vergleich zu gesunden Freiwilligen vermuten.
Kinder und Jugendliche
Die Analyse des pharmakokinetischen Profils von Sildenafil bei den in klinische Studien mit Kindern eingeschlossenen Patienten zeigte, dass das Körpergewicht eine gute Vorhersage der Arzneimittelexposition bei Kindern erlaubt. Bei einem Körpergewicht von 10 bis 70 kg beträgt die Plasmahalbwertzeit von Sildenafil 4,2 bis 4,4 Stunden und zeigt dabei keine Unterschiede, die man als klinisch relevant einstufen könnte. Bei Patienten mit einem Gewicht von 70, 20 und 10 kg betrug die Cmax nach einer oralen Einzeldosis von 20 mg Sildenafil 49, 104 und 165 ng/ml. Bei Patienten mit einem Gewicht von 70, 20 und 10 kg betrug die Cmax nach einer oralen Einzeldosis von 10 mg Sildenafil 24, 53 und 85 ng/ml. Die tmax betrug etwa 1 Stunde und war nahezu unabhängig vom Körpergewicht.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität und zum kanzerogenen Potenzial sowie zur Reproduktions- und zur Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Bei Jungtieren von Ratten, die prä- und postnatal mit 60 mg/kg Sildenafil behandelt worden waren, zeigten sich bei einer Exposition, die etwa dem 50-Fachen der erwarteten Exposition beim Menschen bei einer Dosis von 20 mg dreimal täglich entsprach, eine verminderte Wurfgröße, ein geringeres Gewicht der Jungtiere an Tag 1 und ein vermindertes Überleben bis zum Tag 4. Effekte in präklinischen Studien wurden bei einer Exposition beobachtet, die so weit über der maximalen Dosis beim Menschen lagen, dass sie als für den klinischen Einsatz nicht relevant erachtet wurden.
Im Tiermodell wurden bei klinisch relevanten Konzentrationen keine Nebenwirkungen mit möglichen Auswirkungen auf die klinische Anwendung beobachtet, die nicht ebenfalls in klinischen Studien aufgetreten sind.
Pulver zur Herstellung einer Suspension zum Einnehmen:
Sorbitol (Ph.Eur.) (E 420)
Citronensäure
Sucralose
Natriumcitrat (E 331)
Xanthangummi
Titandioxid (E 171)
Natriumbenzoat (E 211)
hochdisperses Siliciumdioxid
Traubenaroma:
Maltodextrin
Traubensaftkonzentrat
arabisches Gummi
Ananassaftkonzentrat
Citronensäure
natürliche Aromastoffe
Nicht zutreffend.
2 Jahre
Nach Rekonstitution ist die Suspension zum Einnehmen für 30 Tage stabil.
Pulver
Nicht über 30 °C lagern.
In der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Suspension zum Einnehmen
Nicht über 30 °C lagern oder im Kühlschrank lagern (2 °C bis 8 °C).
Nicht einfrieren.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
Eine 125-ml-Braunglasflasche (mit einem Schraubdeckel aus Polypropylen) enthält 32,27 g Pulver zur Herstellung einer Suspension zum Einnehmen.
Nach Rekonstitution enthält die Flasche 112 ml Suspension zum Einnehmen, von denen 90 ml zur Dosierung und Einnahme vorgesehen sind.
Packungsgröße: 1 Flasche
Jede Packung enthält weiterhin einen Messbecher aus Polypropylen (mit Graduierung bei 30 ml), eine 3-ml-Applikationsspritze für Zubereitungen zum Einnehmen aus Polypropylen mit einem HDPE-Kolben und einen Flaschenadapter zum Aufstecken aus LDPE.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Es wird empfohlen, dass ein Apotheker die Revatio Suspension zum Einnehmen vor der Abgabe an den Patienten rekonstituiert.
Anweisung zur Rekonstitution
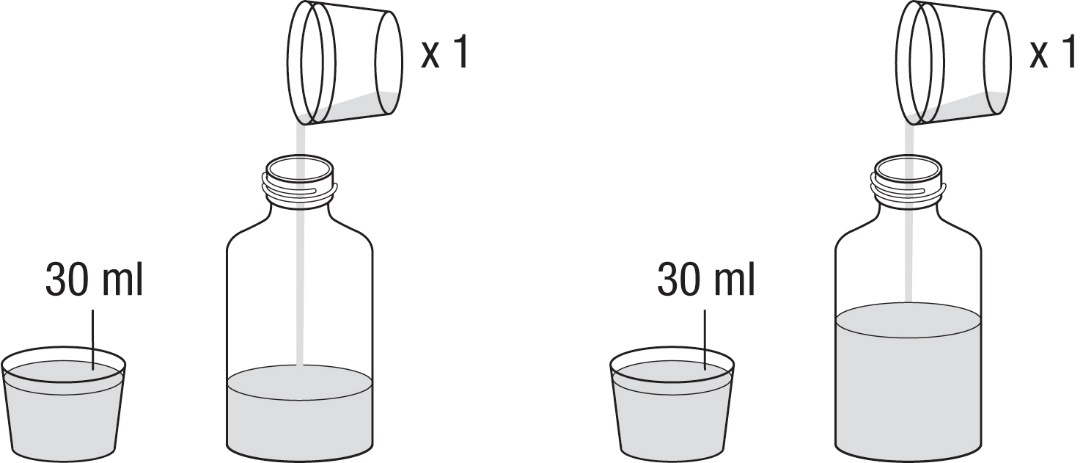
Hinweis: Um den Inhalt der Flasche zu rekonstituieren, sollte unabhängig von der einzunehmenden Dosis immer ein Gesamtvolumen von 90 ml Wasser (3 x 30 ml) verwendet werden.
1.Klopfen Sie leicht an die Flasche, damit sich das Pulver lockert.
2.Entfernen Sie den Verschluss.
3.Messen Sie 30 ml Wasser ab, indem Sie den Messbecher (der der Packung beiliegt) bis zur
Markierungslinie auffüllen, und geben Sie das Wasser in die Flasche. Messen Sie mit dem Becher
nochmal 30 ml Wasser ab und geben Sie dieses ebenfalls in die Flasche (Abbildung 1).
|
Abbildung 1 |
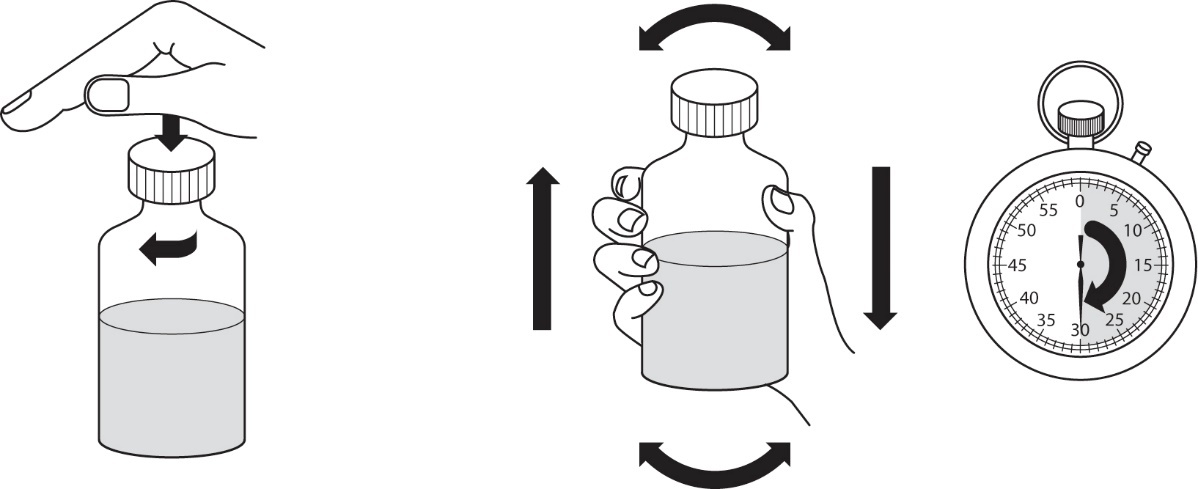
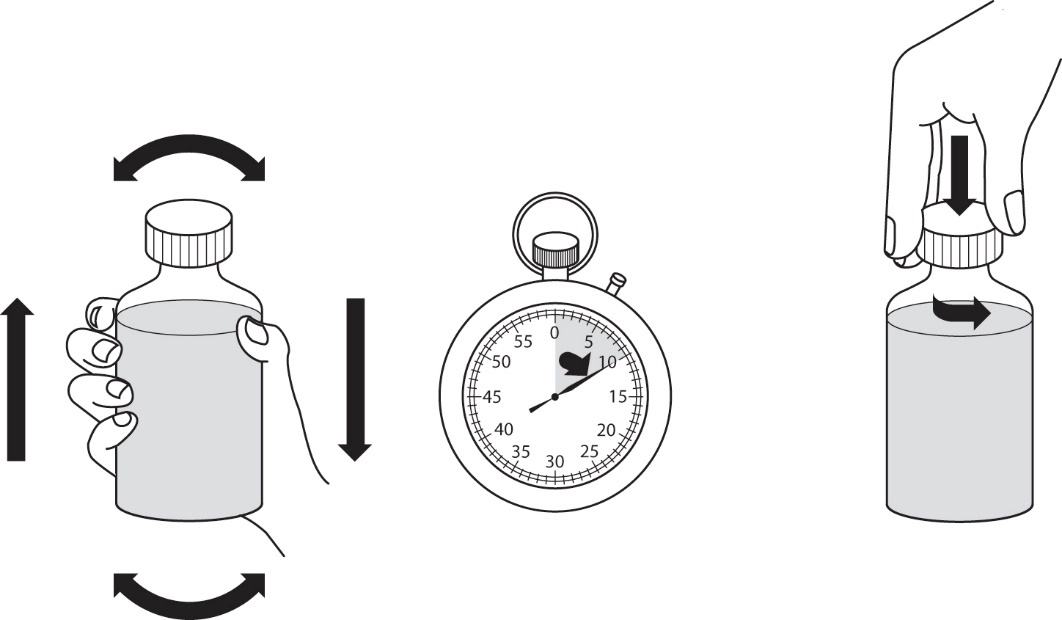
4.Schrauben Sie den Verschluss wieder auf und schütteln Sie die Flasche kräftig für mindestens 30 Sekunden (Abbildung 2).
|
Abbildung 2 |
5.Entfernen Sie den Verschluss.
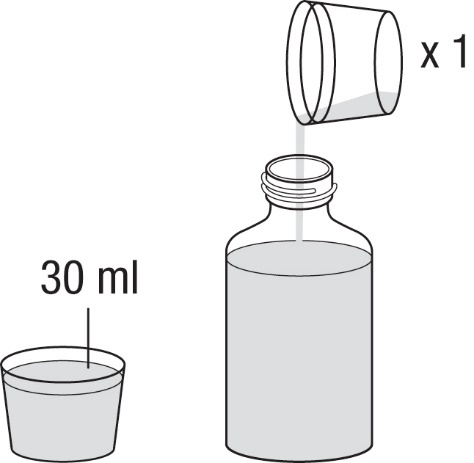
6.Messen Sie mit dem Messbecher nochmals 30 ml Wasser ab und geben Sie es in die Flasche. Unabhängig von der von Ihnen einzunehmenden Dosis sollten Sie immer ein Gesamtvolumen von 90 ml Wasser (3 x 30 ml) zugeben (Abbildung 3).
|
Abbildung 3 |
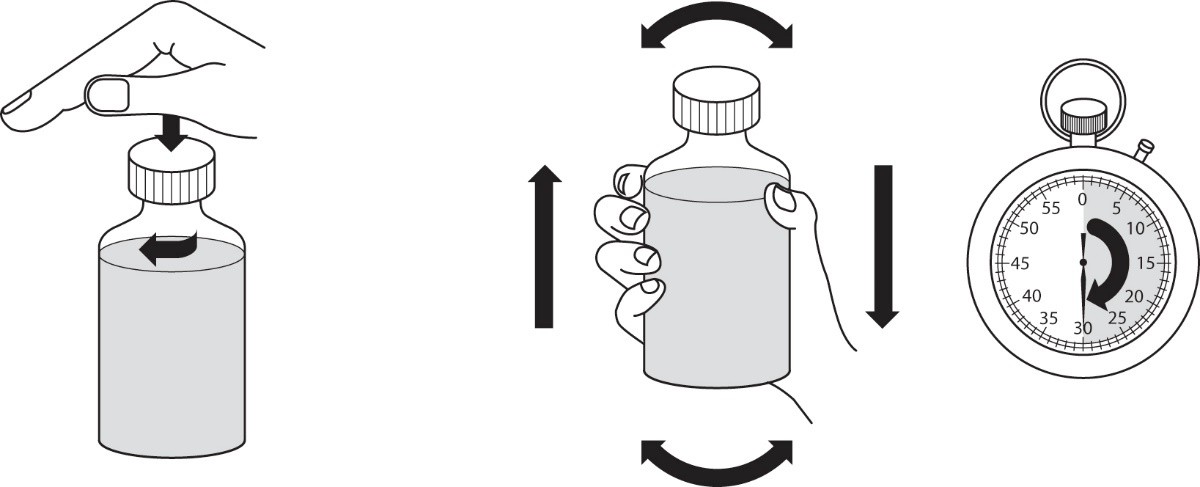
7.Schrauben Sie den Verschluss wieder auf und schütteln Sie die Flasche kräftig für mindestens 30 Sekunden (Abbildung 4).
|
Abbildung 4 |
8.Entfernen Sie den Verschluss.
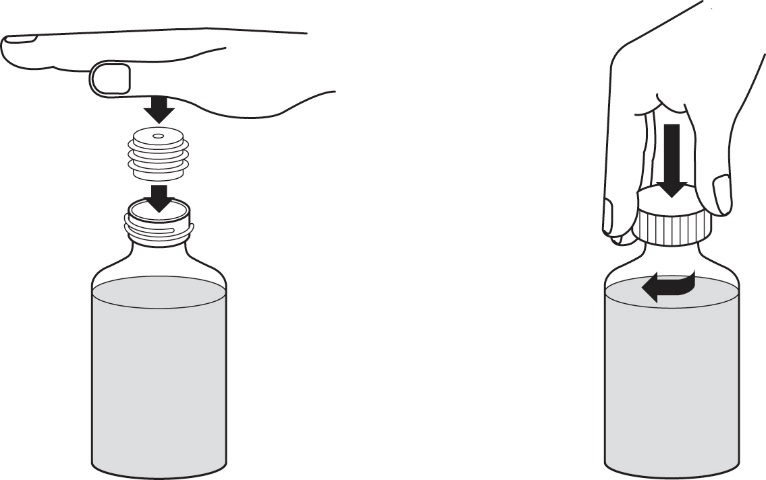
9.Drücken Sie den Flaschenadapter in den Flaschenhals wie in Abbildung 5 gezeigt. Der Flaschenadapter wird benötigt, damit Sie die Applikationsspritze für Zubereitungen zum Einnehmen mit dem Arzneimittel aus der Flasche befüllen können. Schrauben Sie dann den Verschluss wieder auf.
|
Abbildung 5 |
10.Nach der Rekonstitution ergibt das Pulver eine weiße Suspension zum Einnehmen mit Traubengeschmack. Schreiben Sie das Verfalldatum der rekonstituierten Suspension zum Einnehmen auf das Flaschenetikett (das Verfalldatum der rekonstituierten Suspension zum Einnehmen ist 30 Tage nach dem Datum der Rekonstitution). Nicht verwendete Suspension zum Einnehmen sollte nach diesem Datum beseitigt oder dem Apotheker zurückgebracht werden.
Anweisung zur Anwendung
1.Schütteln Sie vor der Anwendung die geschlossene Flasche mit der rekonstituierten Suspension zum Einnehmen kräftig für mindestens 10 Sekunden. Entfernen Sie den Verschluss (Abbildung 6).
|
Abbildung 6 |
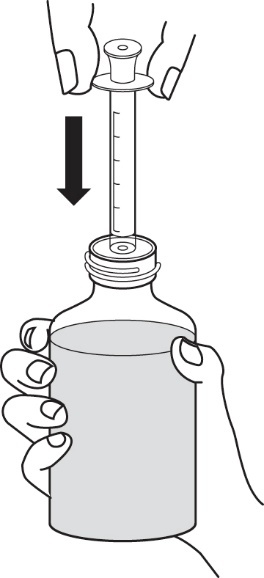
2.Stellen Sie die Flasche aufrecht auf eine ebene Fläche und stecken Sie die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen in den Adapter (Abbildung 7).
|
Abbildung 7 |
3.Stellen Sie die Flasche auf den Kopf und halten Sie die Applikationsspritze für Zubereitungen zum Einnehmen fest. Ziehen Sie den Kolben der Applikationsspritze für Zubereitungen zum Einnehmen langsam bis zu der Markierung heraus, die Ihrer Dosis entspricht (die Entnahme von 1 ml entspricht einer Dosis von 10 mg, die Entnahme von 2 ml entspricht einer Dosis von 20 mg). Für eine genaue Abmessung der Dosis sollte sich die obere Kante des Kolbens auf gleicher Höhe wie die entsprechende Markierung auf der Applikationsspritze für Zubereitungen zum Einnehmen befinden (Abbildung 8).
|
Abbildung 8 |
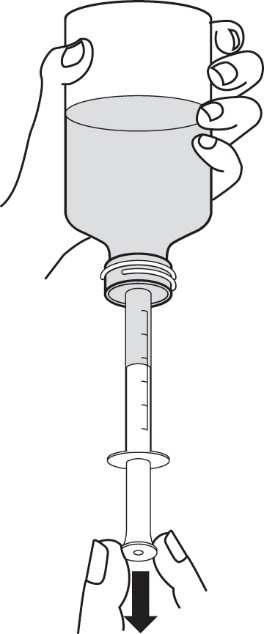
4.Falls große Luftblasen auftreten, schieben Sie den Kolben wieder langsam in die Spritze zurück. Dadurch wird das Arzneimittel wieder in die Flasche zurück gedrückt. Wiederholen Sie dann nochmals den Vorgang ab Schritt 3.
5.Drehen Sie anschließend die Flasche mit aufgesteckter Applikationsspritze für Zubereitungen zum Einnehmen wieder zurück in die aufrechte Position. Entfernen Sie dann die Applikationsspritze für Zubereitungen zum Einnehmen von der Flasche.
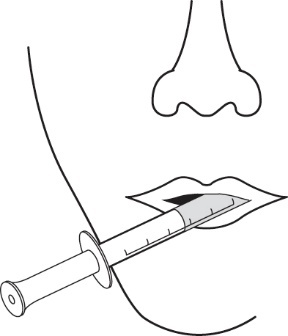
6.Stecken Sie die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen in den Mund. Richten Sie die Spitze der Applikationsspritze für Zubereitungen zum Einnehmen gegen die Innenseite der Wange. Drücken Sie dann den Kolben LANGSAM in die Applikationsspritze für Zubereitungen zum Einnehmen hinein. Pressen Sie das Arzneimittel nicht zu schnell heraus. Wenn das Arzneimittel einem Kind gegeben werden soll, achten Sie darauf, dass das Kind vor der Arzneimittelgabe aufrecht sitzt oder aufrecht gehalten wird (Abbildung 9).
|
Abbildung 9 |
7.Schrauben Sie dann den Verschluss wieder auf die Flasche, aber lassen Sie den Flaschenadapter an Ort und Stelle. Waschen Sie die Applikationsspritze für Zubereitungen zum Einnehmen wie nachfolgend beschrieben aus.
Säuberung und Aufbewahrung der Spritze:
1. Die Spritze sollte nach jeder Anwendung ausgewaschen werden. Ziehen Sie dazu den Kolben aus der Spritze heraus und reinigen Sie beide Teile mit Wasser.
2. Trocknen Sie beide Teile ab. Stecken Sie dann den Kolben wieder zurück in die Spritze. Bewahren Sie diese zusammen mit dem Arzneimittel an einem sauberen und sicheren Ort auf.
Nach der Rekonstitution sollte die Suspension zum Einnehmen nur mit der jeder Packung beiliegenden Applikationsspritze für Zubereitungen zum Einnehmen angewendet werden. Für ausführlichere Angaben zur Anwendung siehe die Gebrauchsinformation für den Patienten.
Upjohn EESV
Rivium Westlaan 142
2909 LD Capelle aan den IJssel
Niederlande
EU/1/05/318/003
Datum der Erteilung der Zulassung: 28. Oktober 2005
Datum der letzten Verlängerung der Zulassung: 23. September 2010
August 2024
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu/ verfügbar.
Verschreibungspflichtig
Verkaufsabgrenzung in Österreich
Rezept- und apothekenpflichtig
Packung mit 1 Flasche (N2)
Packungsgrößen in Österreich
Packung mit 1 Flasche
Viatris Healthcare GmbH
Lütticher Straße 5
53842 Troisdorf
Tel.: +49 (0) 800 0700 800
spcde-4v24ro-pv-0