
Steglujan® 5 mg/100 mg Filmtabletten
Steglujan® 15 mg/100 mg Filmtabletten
Steglujan 5 mg/100 mg Filmtabletten
Jede Tablette enthält Ertugliflozin-Pidolsäure, entsprechend 5 mg Ertugliflozin und Sitagliptinphosphat-Monohydrat, entsprechend 100 mg Sitagliptin.
Steglujan 15 mg/100 mg Filmtabletten
Jede Tablette enthält Ertugliflozin-Pidolsäure, entsprechend 15 mg Ertugliflozin und Sitagliptinphosphat-Monohydrat, entsprechend 100 mg Sitagliptin.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette)
Steglujan 5 mg/100 mg Filmtabletten
Beige, 12 x 7,4 mm große, ovale Filmtabletten mit der Prägung „554“ auf einer Seite und glatt auf der anderen Seite.
Steglujan 15 mg/100 mg Filmtabletten
Braune, 12 x 7,4 mm große, ovale Filmtabletten mit der Prägung „555“ auf einer Seite und glatt auf der anderen Seite.
Steglujan ist bei Erwachsenen ab 18 Jahren mit Typ‑2 Diabetes mellitus zusätzlich zu Diät und Bewegung angezeigt:
zur Verbesserung der Blutzuckerkontrolle bei Patienten, deren Blutzucker unter Metformin und/oder einem Sulfonylharnstoff und einem der in Steglujan enthaltenen Einzelwirkstoffe nicht ausreichend gesenkt werden kann.
bei Patienten, die bereits mit der Kombination aus Ertugliflozin und Sitagliptin in Form von einzelnen Tabletten behandelt werden.
(Zu Studienergebnissen für die Kombinationen und die Wirkung auf die Blutzuckerkontrolle, siehe Abschnitte 4.4, 4.5 und 5.1.)
Dosierung
Die empfohlene Anfangsdosis beträgt 5 mg Ertugliflozin/100 mg Sitagliptin einmal täglich. Sofern eine zusätzliche Blutzuckersenkung notwendig ist, kann die Dosis bei Patienten, welche die Anfangsdosis vertragen, auf 15 mg Ertugliflozin/100 mg Sitagliptin einmal täglich erhöht werden.
Bei Patienten, die mit Ertugliflozin behandelt werden und auf Steglujan umgestellt werden, kann die Ertugliflozin-Dosis beibehalten werden.
Wenn Steglujan in Kombination mit Insulin oder mit einem Insulin-Sekretagogum (einem Arzneimittel zur Anregung der Insulinsekretion) angewendet wird, kann es notwendig sein, die Dosis des Insulins oder des Insulin-Sekretagogums zu verringern, um das Risiko einer Hypoglykämie zu reduzieren (siehe Abschnitte 4.4, 4.5 und 4.8).
Bei Patienten mit einer Hypovolämie wird empfohlen, diese vor Beginn der Behandlung mit Steglujan entsprechend zu korrigieren (siehe Abschnitt 4.4).
Ausgelassene Dosis
Falls eine Einnahme vergessen wurde, sollte diese nachgeholt werden, sobald der Patient daran denkt. Die Patienten sollten nicht die doppelte Dosis Steglujan am selben Tag einnehmen.
Besondere Patientengruppen
Eingeschränkte Nierenfunktion
Die Überprüfung der Nierenfunktion wird vor Beginn der Behandlung mit Steglujan und in regelmäßigen Abständen danach empfohlen (siehe Abschnitt 4.4).
Der Beginn einer Behandlung mit diesem Arzneimittel wird bei Patienten mit einer geschätzten glomerulären Filtrationsrate (eGFR) unter 45 ml/min/1,73 m2 oder einer Kreatinin-Clearance (CrCl) unter 45 ml/min nicht empfohlen (siehe Abschnitt 4.4).
Bei Patienten mit einer eGFR ≥ 45 bis < 60 ml/min/1,73 m2 sollte eine Steglujan Behandlung mit 5 mg/100 mg begonnen und zur Blutzuckerkontrolle bei Bedarf auf 15 mg/100 mg hochtitriert werden.
Da die blutzuckersenkende Wirkung von Ertugliflozin bei Patienten mit moderater Einschränkung der Nierenfunktion verringert und bei Patienten mit schwerer Einschränkung der Nierenfunktion wahrscheinlich nicht vorhanden ist, sollte, falls eine weitere Blutzuckerkontrolle erforderlich ist, die zusätzliche Gabe anderer antihyperglykämischer Arzneimittel in Betracht gezogen werden (siehe Abschnitt 4.4).
Bei einer eGFR anhaltend unter 45 ml/min/1,73 m2 oder einer CrCl anhaltend unter 45 ml/min sollte die Behandlung mit Steglujan abgebrochen werden.
Die Fixdosiskombination aus Ertugliflozin und Sitagliptin sollte bei Patienten mit schwerer Einschränkung der Nierenfunktion, mit terminaler Niereninsuffizienz (end stage renal disease, ESRD) oder bei dialysepflichtigen Patienten nicht angewendet werden, da keine klinischen Daten vorliegen, welche die Wirksamkeit bei diesen Patienten belegen.
Eingeschränkte Leberfunktion
Bei Patienten mit leichter oder moderater Einschränkung der Leberfunktion ist eine Dosisanpassung für Steglujan nicht erforderlich. Die Anwendung von Steglujan wurde bei Patienten mit schwerer Einschränkung der Leberfunktion nicht untersucht und wird für diese Patienten nicht empfohlen (siehe Abschnitt 5.2).
Ältere Patienten
Eine altersabhängige Dosisanpassung für Steglujan wird nicht empfohlen. Die Wahrscheinlichkeit einer eingeschränkten Nierenfunktion ist bei älteren Patienten höher. Da Abweichungen der Nierenfunktion nach Beginn der Behandlung mit Ertugliflozin auftreten können und Sitagliptin bekanntermaßen vornehmlich über die Nieren ausgeschieden wird, sollte die Nierenfunktion bei älteren Patienten häufiger kontrolliert werden. Die Nierenfunktion und das Risiko einer Hypovolämie sollten beachtet werden (siehe Abschnitte 4.4 und 4.8).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Steglujan bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Steglujan sollte einmal täglich am Morgen unabhängig von den Mahlzeiten eingenommen werden. Bei Schluckbeschwerden kann die Tablette zerteilt oder zermahlen werden, da es sich um eine Darreichungsform mit sofortiger Wirkstofffreisetzung handelt.
Überempfindlichkeit gegen die Wirkstoffe oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Allgemeines
Steglujan sollte nicht bei Patienten mit Typ‑1 Diabetes mellitus angewendet werden. Es kann das Risiko einer diabetischen Ketoazidose (DKA) bei diesen Patienten erhöhen.
Akute Pankreatitis
Die Anwendung von Dipeptidylpeptidase‑4 (DPP‑4) Inhibitoren wurde mit einem Risiko für die Entwicklung einer akuten Pankreatitis assoziiert. Die Patienten sollten informiert werden, dass anhaltende starke Bauchschmerzen das charakteristische Symptom einer akuten Pankreatitis sein können. Nach Absetzen von Sitagliptin wurde ein Abklingen der Pankreatitis beobachtet (mit oder ohne supportive Behandlung), aber es wurde auch über sehr seltene schwerwiegende Fälle nekrotisierender oder hämorrhagischer Pankreatitis und/oder Todesfälle berichtet. Bei Verdacht auf eine Pankreatitis sind Steglujan sowie andere potenziell eine Pankreatitis verursachende Arzneimittel abzusetzen; im Falle der Bestätigung einer akuten Pankreatitis ist die Therapie mit Steglujan nicht wieder aufzunehmen. Bei Patienten mit einer Pankreatitis in der Krankengeschichte ist Vorsicht geboten.
Hypotonie/Hypovolämie
Ertugliflozin verursacht eine osmotische Diurese, die zu einem intravaskulären Volumenmangel führen kann. Demzufolge kann eine symptomatische Hypotonie nach Beginn der Behandlung mit Steglujan (siehe Abschnitt 4.8), insbesondere bei Patienten mit eingeschränkter Nierenfunktion (eGFR unter 60 ml/min/1,73 m2 oder CrCl unter 60 ml/min), bei älteren Patienten (≥ 65 Jahre), bei Patienten, die Diuretika einnehmen, oder bei Patienten unter Antihypertonika mit einer Hypotonie in der Vorgeschichte, auftreten. Vor Beginn der Behandlung mit Steglujan sollte der Volumenstatus überprüft und, sofern angezeigt, korrigiert werden. Die Patienten sind auf entsprechende Anzeichen und Symptome einer Hypotonie/Hypovolämie nach Therapiebeginn zu überwachen.
Aufgrund des Wirkmechanismus induziert Ertugliflozin eine osmotische Diurese und führt zu einem Anstieg des Serumkreatinins und einer Abnahme der eGFR. Anstieg des Serumkreatinins und Abnahme der eGFR waren bei Patienten mit moderater Einschränkung der Nierenfunktion stärker ausgeprägt (siehe Abschnitt 4.8).
Im Fall von Erkrankungen, die zu einem Flüssigkeitsverlust führen können (z. B. gastrointestinale Erkrankungen), wird empfohlen, die Patienten, die Steglujan erhalten, sorgfältig hinsichtlich ihres Volumenstatus (z. B. körperliche Untersuchung, Blutdruckmessung, Laboruntersuchungen einschließlich Bestimmung des Hämatokriten) und ihrer Elektrolytwerte zu überwachen. Eine vorübergehende Unterbrechung der Behandlung mit Steglujan sollte in Betracht gezogen werden, bis der Flüssigkeitsverlust korrigiert wurde.
Diabetische Ketoazidose
Seltene Fälle von DKA, einschließlich lebensbedrohlicher und tödlicher Fälle, wurden in klinischen Studien und nach Markteinführung bei Patienten berichtet, die eine Behandlung mit Natrium-Glucose-Co-Transporter 2(SGLT2)-Inhibitoren erhielten, einschließlich Ertugliflozin. In einer Reihe von Fällen zeigte sich ein untypisches Krankheitsbild mit nur mäßig erhöhtem Blutzuckerspiegel unter 14 mmol/l (250 mg/dl). Ob eine DKA mit größerer Wahrscheinlichkeit bei höheren Dosen von Ertugliflozin auftritt, ist nicht bekannt.
Das Risiko einer DKA muss beim Auftreten von unspezifischen Symptomen wie Übelkeit, Erbrechen, Anorexie, Bauchschmerzen, übermäßigem Durst, Schwierigkeiten beim Atmen, Verwirrtheit, ungewöhnlicher Müdigkeit oder Schläfrigkeit in Betracht gezogen werden. Unabhängig vom Blutzuckerspiegel sollten Patienten beim Auftreten dieser Symptome unverzüglich auf eine Ketoazidose hin untersucht werden.
Bei Patienten, bei denen ein Verdacht auf eine DKA besteht oder eine DKA diagnostiziert wurde, ist die Behandlung mit Steglujan sofort abzusetzen.
Die Behandlung sollte bei Patienten unterbrochen werden, die aufgrund größerer chirurgischer Eingriffe oder akuter schwerer Erkrankungen in ein Krankenhaus kommen. Bei diesen Patienten wird eine Überwachung der Ketonkörperkonzentration empfohlen. Die Ermittlung der Ketonkörperkonzentration im Blut ist der Ermittlung der Konzentration im Urin vorzuziehen. Die Behandlung mit Steglujan kann erneut aufgenommen werden, wenn die Ketonkörperkonzentration normal ist und der Zustand des Patienten sich stabilisiert hat.
Vor Beginn einer Behandlung mit Steglujan sind Faktoren in der Anamnese des Patienten, die ihn für eine Ketoazidose prädisponieren könnten, abzuwägen.
Zu den Patienten, für die ein erhöhtes Risiko einer DKA bestehen könnte, gehören Patienten mit einer geringen Funktionsreserve der Beta-Zellen (z. B. Patienten mit Typ‑2 Diabetes und niedrigem C‑Peptid oder latentem Autoimmundiabetes bei Erwachsenen [LADA] oder Patienten mit anamnestisch bekannter Pankreatitis), Patienten mit Erkrankungen, die zu eingeschränkter Nahrungsaufnahme oder schwerer Dehydratation führen, Patienten, bei denen die Insulindosis herabgesetzt wird, und Patienten mit erhöhtem Insulinbedarf infolge einer akuten Krankheit, einer Operation oder Alkoholmissbrauchs. Bei diesen Patienten sind SGLT2-Inhibitoren mit Vorsicht anzuwenden.
Die Wiederaufnahme der Behandlung mit einem SGLT2-Inhibitor wird bei Patienten nicht empfohlen, die unter der Behandlung mit einem SGLT2-Inhibitor zuvor eine DKA entwickelt hatten, es sei denn, es wurde ein anderer eindeutiger auslösender Faktor ermittelt und beseitigt.
Die Sicherheit und Wirksamkeit von Steglujan bei Patienten mit Typ‑1 Diabetes ist bisher nicht belegt und Steglujan sollte nicht zur Behandlung von Patienten mit Typ‑1 Diabetes angewendet werden. Auf der Grundlage begrenzter Daten aus klinischen Studien scheint eine DKA häufig aufzutreten, wenn Patienten mit Typ‑1 Diabetes mit SGLT2-Inhibitoren behandelt werden.
Amputationen der unteren Gliedmaßen
Im Rahmen der Langzeitstudie zur Untersuchung der kardiovaskulären Endpunkte (cardiovasular outcome) VERTIS-CV (eValuation of ERTugliflozin effIcacy and Safety, CardioVascular), einer Studie bei Patienten mit Typ-2 Diabetes mellitus und bestehender atherosklerotischer kardiovaskulärer Erkrankung, wurden hinsichtlich nicht-traumatischer Amputationen der unteren Gliedmaßen (in erster Linie von Zehen) mit einer Inzidenz von 2 % (0,57 Ereignisse pro 100 Patientenjahre), 2,1 % (0,60 Ereignisse pro 100 Patientenjahre) bzw. 1,6 % (0,47 Ereignisse pro 100 Patientenjahre) für die Ertugliflozin 5 mg-, Ertugliflozin 15 mg- bzw. Placebogruppen beobachtet. Die Ereignisraten für Amputationen der unteren Gliedmaßen waren 0,75 bzw. 0,96 versus 0,74 Ereignisse pro 100 Patientenjahre für Ertugliflozin 5 mg bzw. Ertugliflozin 15 mg versus Placebo. Eine erhöhte Anzahl von Amputationen der unteren Gliedmaßen (in erster Linie von Zehen) ist in klinischen Langzeitstudien bei Patienten mit Typ-2 Diabetes mellitus mit SGLT2-Inhibitoren beobachtet worden. Es ist nicht bekannt, ob dies einen Klasseneffekt darstellt. Daher ist es wichtig, Patienten mit Diabetes eine routinemäßige vorbeugende Fußpflege zu empfehlen.
Eingeschränkte Nierenfunktion
Die Wirksamkeit von Ertugliflozin hinsichtlich der Blutzuckerkontrolle hängt von der Nierenfunktion ab, sodass die glykämische Wirksamkeit bei Patienten mit moderater Einschränkung der Nierenfunktion verringert ist, und wahrscheinlich bei Patienten mit schwerer Einschränkung der Nierenfunktion ausbleibt (siehe Abschnitt 4.2).
Der Beginn einer Behandlung mit Steglujan wird bei Patienten mit einer eGFR unter 45 ml/min/1,73 m2 oder einer CrCl unter 45 ml/min nicht empfohlen. Bei einer eGFR anhaltend unter 45 ml/min/1,73 m2 oder einer CrCl anhaltend unter 45 ml/min sollte die Behandlung mit Steglujan aufgrund einer verringerten Wirksamkeit abgebrochen werden.
Die Überwachung der Nierenfunktion wird wie folgt empfohlen:
Vor Beginn der Behandlung mit Steglujan und in regelmäßigen Abständen während der Behandlung (siehe Abschnitt 4.2).
Häufiger bei Patienten mit einer eGFR unter 60 ml/min/1,73 m2 oder einer CrCl unter 60 ml/min.
Hypoglykämie bei gemeinsamer Anwendung mit Insulin und Insulin-Sekretagoga
Ertugliflozin kann das Risiko einer Hypoglykämie erhöhen, wenn es gemeinsam mit Insulin und/oder einem Insulin-Sekretagogum angewendet wird, da diese bekanntlich Hypoglykämien verursachen (siehe Abschnitt 4.8). Bei der Anwendung von Sitagliptin in Kombination mit Insulin oder einem Sulfonylharnstoff wurden Hypoglykämien beobachtet. Demzufolge kann es bei gemeinsamer Anwendung mit Steglujan notwendig sein, die Dosis des Insulins oder des Insulin-Sekretagogums zu verringern, um das Risiko für eine Hypoglykämie zu minimieren (siehe Abschnitte 4.2 und 4.5).
Genitale Pilzinfektionen
Ertugliflozin erhöht das Risiko für genitale Pilzinfektionen. In klinischen Studien mit SGLT2-Inhibitoren kam es bei Patienten mit Pilzinfektionen in der Vorgeschichte und bei Männern ohne Beschneidung mit höherer Wahrscheinlichkeit zu genitalen Pilzinfektionen (siehe Abschnitt 4.8). Diese Patienten sollten entsprechend überwacht und ggf. behandelt werden.
Harnwegsinfektionen
Die Glucoseausscheidung über den Urin kann mit einem erhöhten Risiko für Harnwegsinfektionen einhergehen (siehe Abschnitt 4.8). Bei der Behandlung einer Pyelonephritis oder einer Urosepsis sollte eine zeitweise Unterbrechung der Behandlung mit Ertugliflozin in Betracht gezogen werden.
Nekrotisierende Fasziitis des Perineums (Fournier-Gangrän)
Nach dem Inverkehrbringen wurden Fälle von nekrotisierender Fasziitis des Perineums (auch als Fournier-Gangrän bezeichnet) bei weiblichen und männlichen Patienten gemeldet, die SGLT2-Hemmer einnahmen. Hierbei handelt es sich um ein seltenes, aber schwerwiegendes und potenziell lebensbedrohliches Ereignis, das einen dringenden chirurgischen Eingriff und eine Behandlung mit Antibiotika erfordert.
Den Patienten sollte empfohlen werden, sich an einen Arzt zu wenden, wenn bei ihnen die Symptome Schmerzen, Berührungsempfindlichkeit, Erythem oder Schwellungen im Bereich der Genitalien oder des Perineums gleichzeitig mit Fieber oder Unwohlsein auftreten. Beachten Sie, dass im Vorfeld einer nekrotisierenden Fasziitis Infektionen des Urogenitaltrakts oder Perinealabszesse auftreten können. Bei Verdacht auf Fournier-Gangrän ist Steglujan abzusetzen und unverzüglich eine Behandlung (u. a. die Gabe von Antibiotika und chirurgisches Debridement) einzuleiten.
Überempfindlichkeitsreaktionen
Nach Markteinführung wurde bei Patienten unter Sitagliptin über schwerwiegende Überempfindlichkeitsreaktionen berichtet (siehe Abschnitt 4.8). Diese Reaktionen schließen Anaphylaxie, Angioödem und exfoliative Hauterscheinungen einschließlich Stevens-Johnson-Syndrom ein. Das Auftreten dieser Reaktionen erfolgte innerhalb der ersten 3 Monate nach Beginn der Behandlung, einigen Berichten zufolge nach der ersten Dosis. Falls ein Verdacht auf eine Überempfindlichkeitsreaktion besteht, ist Steglujan abzusetzen. Andere mögliche Ursachen für das Ereignis sind abzuklären und eine alternative Diabetesbehandlung ist einzuleiten.
Bullöses Pemphigoid
Nach Markteinführung wurde bei Patienten unter DPP‑4-Inhibitoren einschließlich Sitagliptin über das Auftreten eines bullösen Pemphigoids berichtet. Falls ein Verdacht auf bullöses Pemphigoid besteht, ist Steglujan abzusetzen.
Ältere Patienten
Ältere Patienten können ein erhöhtes Risiko für eine Hypovolämie und eine eingeschränkte Nierenfunktion haben. Patienten über 65 Jahre, die mit Ertugliflozin behandelt wurden, hatten eine höhere Inzidenz für Nebenwirkungen im Zusammenhang mit einer Hypovolämie als jüngere Patienten. In der Langzeitstudie zur Untersuchung der kardiovaskulären Endpunkte VERTIS-CV waren Sicherheit und Wirksamkeit ähnlich bei Patienten, die 65 Jahre oder älter waren, verglichen mit Patienten unter 65 Jahren (siehe Abschnitte 4.2 und 4.8).
Herzinsuffizienz
Es liegen keine Erfahrungen aus klinischen Studien mit Ertugliflozin bei Patienten mit Herzinsuffizienz der New York Heart Association (NYHA) Klasse IV vor.
Urin-Laboruntersuchungen
Aufgrund des Wirkmechanismus von Ertugliflozin fallen Urintests auf Glucose bei mit Steglujan behandelten Patienten positiv aus. Zur Blutzuckerkontrolle sollten andere Methoden angewendet werden.
Einfluss auf den 1,5-Anhydroglucitol (1,5‑AG) Assay
Die Überwachung der Blutzuckerkontrolle mit Hilfe des 1,5‑AG Assays wird aufgrund unzuverlässiger Messergebnisse des 1,5‑AG Assays bei Patienten, die Arzneimittel einnehmen, die SGLT2-Inhibitoren enthalten, nicht empfohlen. Zur Blutzuckerkontrolle sollten andere Methoden angewendet werden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tablette, d. h., es ist nahezu „natriumfrei“.
Es wurden keine pharmakokinetischen Arzneimittelwechselwirkungsstudien mit Steglujan durchgeführt. Diese Studien wurden jedoch mit Ertugliflozin und Sitagliptin, den in Steglujan enthaltenen Einzelwirkstoffen, durchgeführt.
Ertugliflozin
Pharmakodynamische Wechselwirkungen
Diuretika
Ertugliflozin kann die diuretische Wirkung von Diuretika verstärken und so das Risiko für eine Dehydratation und Hypotonie erhöhen (siehe Abschnitt 4.4).
Insulin und Insulin-Sekretagoga
Insulin und Insulin-Sekretagoga wie Sulfonylharnstoffe verursachen Hypoglykämien. Ertugliflozin kann das Risiko für eine Hypoglykämie erhöhen, sofern es gemeinsam mit Insulin oder einem Insulin-Sekretagogum angewendet wird. Demzufolge kann es bei gemeinsamer Anwendung mit Steglujan notwendig sein, die Dosis des Insulins oder des Insulin-Sekretagogums zu verringern, um das Risiko für eine Hypoglykämie zu reduzieren (siehe Abschnitte 4.2, 4.4 und 4.8).
Pharmakokinetische Wechselwirkungen
Wirkungen anderer Arzneimittel auf die Pharmakokinetik von Ertugliflozin
Ertugliflozin wird hauptsächlich mittels Metabolisierung durch UGT1A9 und UGT2B7 ausgeschieden.
Arzneimittelwechselwirkungsstudien bei gesunden Probanden deuten darauf hin, dass die Pharmakokinetik von Ertugliflozin durch Sitagliptin, Metformin, Glimepirid oder Simvastatin nach Einmalgabe nicht beeinflusst wird.
Die mehrfache Gabe von Rifampicin (ein Uridin-5´-trihydrogendiphosphat-Glukuronosyltransferase [UGT]- und Cytochrom P450 [CYP]-Induktor) führt zu einer Reduktion der Fläche unter der Konzentrations-Zeit-Kurve (Area under the curve, AUC) und der maximalen Plasmakonzentration (Cmax) von Ertugliflozin um 39 % bzw. 15 %. Diese Reduktion der Exposition wird als klinisch nicht relevant erachtet, sodass keine Dosisanpassung empfohlen wird. Ein klinisch relevanter Einfluss durch andere Enzyminduktoren (z. B. Carbamazepin, Phenytoin, Phenobarbital) ist nicht zu erwarten.
Der Einfluss von UGT-Inhibitoren auf die Pharmakokinetik von Ertugliflozin wurde klinisch nicht untersucht. Ein möglicher Anstieg der Exposition von Ertugliflozin durch Hemmung von UGT wird nicht als klinisch relevant angesehen.
Wirkungen von Ertugliflozin auf die Pharmakokinetik anderer Arzneimittel
Arzneimittelwechselwirkungsstudien bei gesunden Probanden deuten darauf hin, dass Ertugliflozin keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Sitagliptin, Metformin und Glimepirid hat.
Die gemeinsame Anwendung von Simvastatin mit Ertugliflozin führte zu einem Anstieg der AUC und Cmax von Simvastatin um 24 % bzw. 19 % und zu einem Anstieg der AUC und Cmax von Simvastatinsäure um 30 % bzw. 16 %. Der Mechanismus für den leichten Anstieg von Simvastatin und Simvastatinsäure ist nicht bekannt und wird nicht durch eine Hemmung vom Organo-Anion-Transporter (OATP) durch Ertugliflozin verursacht. Diese Anstiege werden als klinisch nicht relevant erachtet.
Sitagliptin
Pharmakokinetische Wechselwirkungen
Wirkungen anderer Arzneimittel auf Sitagliptin
Sitagliptin wird vorwiegend unverändert über den Urin eliminiert, seine Metabolisierung spielt eine untergeordnete Rolle. In vitro Studien deuten darauf hin, dass CYP3A4, unter Beteiligung von CYP2C8, das hauptverantwortliche Enzym für die begrenzte Metabolisierung von Sitagliptin ist.
Die Metabolisierung könnte jedoch bei schwerer Einschränkung der Nierenfunktion oder bei ESRD eine wichtigere Rolle bei der Ausscheidung von Sitagliptin spielen. Daher ist es möglich, dass potente CYP3A4-Inhibitoren (z. B. Ketoconazol, Itraconazol, Ritonavir, Clarithromycin) die Pharmakokinetik von Sitagliptin bei Patienten mit schwerer Einschränkung der Nierenfunktion oder mit terminaler Niereninsuffizienz verändern.
Arzneimittelwechselwirkungsstudien bei Patienten mit Typ‑2 Diabetes oder bei gesunden Probanden deuten darauf hin, dass Metformin und Ciclosporin keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Sitagliptin haben.
Wirkungen von Sitagliptin auf andere Arzneimittel
In Arzneimittelwechselwirkungsstudien hatte Sitagliptin keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Metformin, Rosiglitazon, Glibenclamid, Simvastatin, Warfarin oder oralen Kontrazeptiva.
Digoxin:
Sitagliptin hatte eine geringe Wirkung auf die Plasmakonzentrationen von Digoxin. Nach Gabe von 0,25 mg Digoxin mit 100 mg Sitagliptin pro Tag über 10 Tage erhöhte sich die Plasma-AUC von Digoxin um durchschnittlich 11 %, die Plasma-Cmax stieg um durchschnittlich 18 % an. Für Digoxin wird keine Dosisanpassung empfohlen. Patienten mit einem Risiko für eine Digoxin-Toxizität sollten jedoch unter einer gemeinsamen Behandlung mit Sitagliptin und Digoxin entsprechend überwacht werden.
Schwangerschaft
Es liegen keine Daten zur Anwendung von Steglujan bei Schwangeren vor. Es liegen begrenzte Daten zur Anwendung von Ertugliflozin bei Schwangeren vor. Basierend auf den Ergebnissen tierexperimenteller Studien kann Ertugliflozin die Entwicklung und Reifung der Nieren beeinflussen (siehe Abschnitt 5.3). Deshalb sollte Steglujan während der Schwangerschaft nicht angewendet werden.
Stillzeit
Es ist nicht bekannt, ob die in Steglujan enthaltenen Einzelwirkstoffe in die Muttermilch übergehen oder ob diese Auswirkungen auf gestillte Neugeborene/Kinder oder auf die Milchbildung haben. Es wurden keine Studien bei laktierenden Tieren mit der in Steglujan enthaltenen Wirkstoffkombination durchgeführt. Ertugliflozin und Sitagliptin gehen in die Milch von laktierenden Ratten über. Ertugliflozin hatte Auswirkungen auf deren Nachkommen. Bei juvenilen Ratten wurden pharmakologisch vermittelte Auswirkungen beobachtet (siehe Abschnitt 5.3). Da die Reifung der Nieren beim Menschen in der Gebärmutter und während der ersten 2 Lebensjahre bei möglicher Exposition durch das Stillen stattfindet, kann ein Risiko für Neugeborene/Kinder nicht ausgeschlossen werden. Steglujan sollte während der Stillzeit nicht angewendet werden.
Fertilität
Die Auswirkung von Steglujan auf die menschliche Fertilität wurde nicht untersucht. In tierexperimentellen Studien wurden keine Auswirkungen von Ertugliflozin oder Sitagliptin auf die Fertilität beobachtet (siehe Abschnitt 5.3).
Steglujan hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Jedoch sollte man bei Fahrtätigkeit oder beim Bedienen von Maschinen beachten, dass unter Sitagliptin über Schwindel und Schläfrigkeit berichtet wurde. Zudem sollten die Patienten darauf aufmerksam gemacht werden, dass bei Anwendung von Steglujan in Kombination mit Insulin oder einem Insulin-Sekretagogum das Risiko für eine Hypoglykämie besteht und das Risiko für Nebenwirkungen im Zusammenhang mit einer Hypovolämie wie z. B. orthostatischer Schwindel erhöht ist (siehe Abschnitte 4.2, 4.4 und 4.8).
Zusammenfassung des Sicherheitsprofils
Ertugliflozin und Sitagliptin
Die Sicherheit von gemeinsam angewendetem Ertugliflozin und Sitagliptin wurde im Rahmen von drei Studien bei insgesamt 990 Patienten mit Typ‑2 Diabetes mellitus über einen Zeitraum von 26 Wochen untersucht: eine faktorielle Studie zur Untersuchung von einmal täglich 5 mg oder 15 mg Ertugliflozin in Kombination mit 100 mg Sitagliptin im Vergleich zu den jeweiligen Einzelwirkstoffen, eine placebokontrollierte Studie zur Untersuchung von einmal täglich 5 mg oder 15 mg Ertugliflozin als Add-on-Therapie zu 100 mg Sitagliptin und Metformin und eine placebokontrollierte Studie zur Untersuchung von einmal täglich 5 mg oder 15 mg Ertugliflozin in Kombination mit 100 mg Sitagliptin als initiale Kombinationstherapie (siehe Abschnitt 5.1). Die Inzidenz und Art der Nebenwirkungen, die im Rahmen dieser drei Studien beobachtet wurden, waren mit den Nebenwirkungen, die unter Behandlung mit den jeweiligen Monotherapien Ertugliflozin und Sitagliptin allein beobachtet wurden, vergleichbar, wie in Tabelle 1 aufgeführt.
Ertugliflozin
Die Sicherheit und Verträglichkeit von Ertugliflozin wurde in 7 placebo- oder aktivkontrollierten Studien bei insgesamt 3 409 Patienten mit Typ-2 Diabetes mellitus untersucht, die mit Ertugliflozin 5 mg oder 15 mg behandelt wurden. Zusätzlich wurde im Rahmen der VERTIS-CV Studie (siehe Abschnitt 5.1) die Sicherheit und Verträglichkeit von Ertugliflozin bei insgesamt 5 493 Patienten mit Typ-2 Diabetes und bestehender atherosklerotischer kardiovaskulärer Erkrankung untersucht, die mit Ertugliflozin 5 mg oder 15 mg über einen mittleren Behandlungszeitraum von 2,9 Jahren behandelt wurden.
Gepoolte placebokontrollierte Studien
Die primäre Sicherheitsbewertung erfolgte anhand von drei gepoolten placebokontrollierten Studien über jeweils 26 Wochen. Ertugliflozin wurde in einer der Studien als Monotherapie und in zwei der Studien als Add-on-Therapie angewendet (siehe Abschnitt 5.1). Diese Daten umfassen die Exposition von 1 029 Patienten mit Ertugliflozin mit einer mittleren Expositionsdauer von ca. 25 Wochen. Die Patienten erhielten 5 mg Ertugliflozin (N = 519), 15 mg Ertugliflozin (N = 510) oder Placebo (N = 515) einmal täglich.
Die am häufigsten berichteten Nebenwirkungen über das klinische Entwicklungsprogramm hinweg waren Harnwegsinfektionen, vulvovaginale Pilzinfektionen und andere genitale Pilzinfektionen bei Frauen. Schwerwiegende DKA traten selten auf (siehe Abschnitt 4.4).
Sitagliptin
Es wurden schwerwiegende Nebenwirkungen einschließlich Pankreatitis und Überempfindlichkeitsreaktionen berichtet. Hypoglykämien wurden in der Kombination mit Sulfonylharnstoffen (4,7 % − 13,8 %) und Insulin (9,6 %) berichtet (siehe Abschnitt 4.4).
Tabellarische Auflistung der Nebenwirkungen
Die unten angegebenen Nebenwirkungen sind gemäß ihrer Häufigkeit und der zugehörigen Systemorganklasse (SOC) eingeteilt, innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach absteigendem Schweregrad aufgeführt. Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1 000, <1/100), selten (≥1/10 000, <1/1 000), sehr selten (<1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 1: Nebenwirkungen aus placebo- und aktivkontrollierten klinischen Studien und Erfahrungen nach Markteinführung
Systemorganklasse | Nebenwirkung |
Infektionen und parasitäre Erkrankungen | |
Sehr häufig | Harnwegsinfektionen†,1 |
Häufig | Candida-Balanitis und andere genitale Pilzinfektionen bei Männern*,†,1 |
Nicht bekannt | Nekrotisierende Fasziitis des Perineums (Fournier-Gangrän)*,a |
Erkrankungen des Blutes und des Lymphsystems | |
Selten | Thrombozytopenie2 |
Erkrankungen des Immunsystems | |
Nicht bekannt | Überempfindlichkeitsreaktionen einschließlich anaphylaktischer Reaktionen*,a,2 |
Stoffwechsel- und Ernährungsstörungen | |
Häufig | Hypoglykämie*,†,1,2 |
Selten | DKA*,†,1 |
Erkrankungen des Nervensystems | |
Häufig | Kopfschmerzen2 |
Gelegentlich | Schwindel2 |
Gefäßerkrankungen | |
Häufig | Hypovolämie*,†,1 |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | |
Nicht bekannt | Interstitielle Lungenkrankheita,2 |
Erkrankungen des Gastrointestinaltrakts | |
Gelegentlich | Obstipation2 |
Nicht bekannt | Letale und nicht letale hämorrhagische und nekrotisierende Pankreatitis*,a,2 |
Erkrankungen der Haut und des Unterhautgewebes | |
Gelegentlich | Pruritusa,2 |
Nicht bekannt | Exfoliative Hauterkrankungen einschließlich Stevens-Johnson-Syndroma,*,2 |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | |
Nicht bekannt | Arthropathiea,2 |
Erkrankungen der Nieren und Harnwege | |
Häufig | Erhöhter Harndrang‡,1 |
Gelegentlich | Dysurie1, Kreatinin im Blut erhöht/glomeruläre Filtrationsrate vermindert†,1 |
Nicht bekannt | Akutes Nierenversagena,2 |
Erkrankungen der Geschlechtsorgane und der Brustdrüse | |
Häufig | Vulvovaginaler Pruritus1 |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | |
Häufig | Durst§,1 |
Untersuchungen | |
Häufig | Serumlipide verändert¶,1, Hämoglobin erhöht**,1, BUN erhöht¶¶,1 |
1 Nebenwirkung unter Ertugliflozin. | |
Beschreibung ausgewählter Nebenwirkungen
Ertugliflozin
Hypovolämie
Ertugliflozin induziert eine osmotische Diurese, die zu einer intravaskulären Volumenkontraktion und zu Nebenwirkungen in Zusammenhang mit einer Hypovolämie führen kann. Auf Datenbasis der gepoolten placebokontrollierten Studien war die Inzidenz von unerwünschten Ereignissen in Zusammenhang mit einer Hypovolämie (Dehydratation, orthostatischer Schwindel, Präsynkope, Synkope, Hypotonie und orthostatische Hypotonie) gering (< 2 %) und zwischen den Patientengruppen unter Ertugliflozin oder Placebo nicht nennenswert unterschiedlich. Anhand des umfangreicheren Datenpools der Phase III Studien zeigte sich in den Subgruppenanalysen, dass Patienten mit einer eGFR < 60 ml/min/1,73 m2, Patienten im Alter von ≥ 65 Jahren und Patienten, die Diuretika einnahmen, in den Ertugliflozingruppen im Vergleich zu den Kontrollgruppen eine höhere Inzidenz für eine Hypovolämie hatten (siehe Abschnitte 4.2 und 4.4). Bei Patienten mit einer eGFR < 60 ml/min/1,73 m2 lag die Inzidenz bei 5,1 %, 2,6 % und 0,5 % in den Gruppen unter 5 mg Ertugliflozin, 15 mg Ertugliflozin bzw. in der Kontrollgruppe. Bei Patienten mit einer eGFR von 45 bis < 60 ml/min/1,73 m2 lag die Inzidenz bei 6,4 %, 3,7 % und 0 % in den Gruppen unter 5 mg Ertugliflozin, 15 mg Ertugliflozin bzw. in der Kontrollgruppe.
Hypoglykämie
Auf Datenbasis der gepoolten placebokontrollierten Studien war die Häufigkeit bestätigter Hypoglykämien bei Patienten unter Ertugliflozin 5 mg und 15 mg (5 % bzw. 4,5 %) im Vergleich zu Placebo (2,9 %) erhöht. In dieser Population betrug die Inzidenz schwerer Hypoglykämien in jeder Subgruppe 0,4 %. Bei Anwendung von Ertugliflozin als Monotherapie lag die Inzidenz von hypoglykämischen Ereignissen in beiden Ertugliflozingruppen bei 2,6 % und in der Placebogruppe bei 0,7 %. Bei Anwendung als Add-on zu Metformin lag die Inzidenz von hypoglykämischen Ereignissen in der Gruppe unter 5 mg Ertugliflozin bei 7,2 %, in der Gruppe unter 15 mg Ertugliflozin bei 7,8 % und in der Placebogruppe bei 4,3 %.
Bei Anwendung von Ertugliflozin oder Sulfonylharnstoff als Add-on zu Metformin war die Inzidenz von Hypoglykämien bei den Patienten unter Sulfonylharnstoff (27 %) höher als im Vergleich zu Ertugliflozin (5,6 % und 8,2 % für Ertugliflozin 5 mg bzw. 15 mg).
Im Rahmen der VERTIS-CV Substudien, bei denen Ertugliflozin als Add-on zu Insulin mit oder ohne Metformin gegeben wurde, lag die Inzidenz einer dokumentierten Hypoglykämie bei den Patienten unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo bei 39,4 %, 38,9 % bzw. 37,5 %. Bei Gabe von Ertugliflozin als Add-on zu Sulfonylharnstoffen lag die Inzidenz für eine Hypoglykämie bei Patienten unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo bei 7,3 %, 9,3 % bzw. 4,2 %. Bei Gabe von Ertugliflozin als Add-on zu Metformin und einem Sulfonylharnstoff lag die Inzidenz für eine Hypoglykämie bei Patienten unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo bei 20 %, 26,5 % bzw. 14,5 %.
Bei Patienten mit moderater Einschränkung der Nierenfunktion, die Insuline, Sulfonylharnstoffe oder Glinide als Hintergrundarzneimittel einnahmen, wurden bestätigte Hypoglykämien bei 36 %, 27 % und 36 % der Patienten unter 5 mg Ertugliflozin, 15 mg Ertugliflozin bzw. Placebo beobachtet (siehe Abschnitte 4.2, 4.4 und 4.5).
Diabetische Ketoazidose
Im Rahmen der VERTIS-CV Studie wurde eine Ketoazidose bei 19 (0,3 %) Patienten unter Ertugliflozin und bei 2 (0,1 %) Patienten unter Placebo festgestellt. Über 7 andere Phase III Studien des klinischen Ertugliflozin-Entwicklungsprogramms hinweg wurde eine Ketoazidose bei 3 (0,1 %) Patienten unter Ertugliflozin und bei 0 (0 %) der Patienten unter Vergleichsmedikation festgestellt (siehe Abschnitt 4.4).
Erhöhtes Kreatinin im Blut/Verminderte glomeruläre Filtrationsrate und renale Ereignisse
Initiale Erhöhungen des mittleren Kreatinins und Abnahmen der mittleren eGFR waren bei Patienten unter Ertugliflozin bei durchgängiger Behandlung im Allgemeinen vorübergehend. Patienten mit moderater Einschränkung der Nierenfunktion zu Studienbeginn hatten größere mittlere Abweichungen, welche bis Woche 26 nicht wieder auf das Ausgangsniveau zurückgingen, sich nach Absetzen der Behandlung aber wieder normalisierten.
Im Rahmen der VERTIS-CV Studie war die Behandlung mit Ertugliflozin mit einer initialen Abnahme der mittleren eGFR verbunden (in Woche 6 −2,7, −3,8 bzw. −0,4 ml/min/1,73 m2 bei Patienten unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo) gefolgt von einer Rückkehr zum Ausgangswert. Im Zusammenhang mit einer kontinuierlichen Langzeitbehandlung mit Ertugliflozin kam es zu einer langsameren Abnahme der mittleren eGFR im Vergleich zu Placebo (über einen Zeitraum von bis zu 260 Wochen).
Im Rahmen der VERTIS-CV Studie lag die Inzidenz von renalen Nebenwirkungen (z. B. akute Nierenschädigung, eingeschränkte Nierenfunktion, akute prärenale Insuffizienz) bei den Patienten in der Gesamtpopulation unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo bei 4,2 %, 4,3 % bzw. 4,7 % und betrug bei den Patienten mit einer eGFR von 30 bis kleiner 60 ml/min/1,73 m2 unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo 9,7 %, 10 % bzw. 10,2 %.
Genitale Pilzinfektionen
Auf Datenbasis der drei gepoolten placebokontrollierten klinischen Studien wurden genitale Pilzinfektionen bei Frauen (z. B. genitale Candidose, genitale Pilzinfektion, Vaginalinfektion, Vulvitis, vulvovaginale Candidose, vulvovaginale Pilzinfektion, Vulvovaginitis) bei 9,1 %, 12 % bzw. 3 % der Patientinnen unter 5 mg Ertugliflozin, 15 mg Ertugliflozin und Placebo beobachtet. Die Behandlung wurde bei Frauen aufgrund von genitalen Pilzinfektionen unter Ertugliflozin und Placebo bei 0,6 % bzw. 0 % der Patientinnen abgebrochen (siehe Abschnitt 4.4).
Auf gleicher Datenbasis lag die Inzidenz von genitalen Pilzinfektionen bei Männern (z. B. Candida-Balanitis, Balanoposthitis, Genitalinfektion, genitale Pilzinfektion) unter 5 mg Ertugliflozin, 15 mg Ertugliflozin und Placebo bei 3,7 %, 4,2 % bzw. 0,4 %. Genitale Pilzinfektionen traten bei Männern ohne Beschneidung häufiger auf. Die Behandlung wurde bei Männern aufgrund von genitalen Pilzinfektionen unter Ertugliflozin und Placebo bei 0,2 % bzw. 0 % der Patienten abgebrochen. In seltenen Fällen wurde eine Phimose berichtet und in manchen Fällen wurde eine Beschneidung durchgeführt (siehe Abschnitt 4.4).
Harnwegsinfektionen
Im Rahmen der VERTIS-CV Studie traten Harnwegsinfektionen in 12,2 %, 12 % bzw. 10,2 % der Patienten unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo auf. Die Inzidenz von schweren Harnwegsinfektionen betrug 0,9 %, 0,4 % bzw. 0,8 % unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo.
Über 7 andere klinische Phase III Studien aus dem Ertugliflozin-Entwicklungsprogramm hinweg betrug die Inzidenz für Harnwegsinfektionen 4 %, 4,1 % bzw. 3,9 % unter Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Placebo. Die meisten Ereignisse waren leicht bis moderat und keine schweren Fälle wurden berichtet.
Sitagliptin
Zusätzlich zu den in der Tabelle oben beschriebenen Nebenwirkungen wurden Nebenwirkungen ungeachtet eines Kausalzusammenhangs mit der Medikation berichtet, wie beispielsweise Infektionen der oberen Atemwege und Nasopharyngitis. Diese traten bei mindestens 5 % der Patienten auf und waren bei Patienten unter Sitagliptin häufiger. Weitere Nebenwirkungen, die ungeachtet eines Kausalzusammenhangs mit der Medikation berichtet wurden, wie beispielsweise Osteoarthrose und Schmerzen in den Gliedmaßen, traten bei Patienten unter Sitagliptin zwar häufiger auf (mit einer Inzidenz von > 0,5 % häufiger unter Sitagliptin als in der Kontrollgruppe), blieben aber unter der 5-%-Schwelle.
Einige Nebenwirkungen wurden häufiger in Studien zur Untersuchung von Sitagliptin in Kombination mit anderen Antidiabetika beobachtet als in Studien zur Untersuchung von Sitagliptin als Monotherapie. Dazu gehörten Hypoglykämien (sehr häufig in Kombination mit Sulfonylharnstoffen und Metformin), Influenza (häufig zusammen mit Insulin [mit oder ohne Metformin]), Übelkeit und Erbrechen (häufig zusammen mit Metformin), Flatulenz (häufig zusammen mit Metformin oder Pioglitazon), Obstipation (häufig in Kombination mit Sulfonylharnstoffen und Metformin), periphere Ödeme (häufig zusammen mit Pioglitazon oder der Kombination von Pioglitazon und Metformin), Somnolenz und Diarrhö (gelegentlich in Kombination mit Metformin) sowie Mundtrockenheit (gelegentlich mit Insulin [mit oder ohne Metformin]).
TECOS („Trial Evaluating Cardiovascular Outcomes with Sitagliptin“)
Die kardiovaskuläre Sicherheitsstudie zu Sitagliptin (TECOS) schloss in der „Intention to Treat Population“ 7 332 Patienten, die mit Sitagliptin 100 mg pro Tag behandelt wurden (oder 50 mg pro Tag, falls die geschätzte glomeruläre Filtrationsrate [eGFR] zu Studienbeginn bei ≥ 30 und
< 50 ml/min/1,73 m2 lag), sowie 7 339 Patienten, die mit Placebo behandelt wurden, ein. Beide Behandlungen wurden zusätzlich zu einer Standardversorgung, die hinsichtlich Hämoglobin A1c (HbA1c)‑Zielwert und kardiovaskulärer (CV) Risikofaktoren den lokalen Therapierichtlinien angepasst war, gegeben. Die Gesamtinzidenz schwerwiegender unerwünschter Ereignisse war bei den Patienten unter Sitagliptin und den Patienten unter Placebo ähnlich.
In der „Intention to Treat Population“ betrug die Inzidenz schwerer Hypoglykämien bei den Patienten, die bei Studieneinschluss Insulin und/oder Sulfonylharnstoff erhielten, 2,7 % unter Behandlung mit Sitagliptin und 2,5 % unter Behandlung mit Placebo. Bei den Patienten, die bei Studieneinschluss weder Insulin noch Sulfonylharnstoff erhielten, betrug die Inzidenz schwerer Hypoglykämien 1 % unter Behandlung mit Sitagliptin und 0,7 % unter Behandlung mit Placebo. Die Inzidenz medizinisch bestätigter Ereignisse von Pankreatitis betrug 0,3 % unter Behandlung mit Sitagliptin und 0,2 % bei Patienten unter Placebo.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: www.bfarm.de, anzuzeigen.
Im Fall einer Überdosierung mit Steglujan sollten die üblichen unterstützenden Maßnahmen (z. B. Elimination von noch nicht aufgenommenem Arzneimittel aus dem Gastrointestinaltrakt, klinische Überwachung des Patienten einschließlich Aufnahme eines Elektrokardiogramms und Einleiten unterstützender Maßnahmen) gemäß dem klinischen Allgemeinzustand des Patienten ergriffen werden.
Ertugliflozin
Bei gesunden Probanden gab es keinerlei Anzeichen von Toxizität bei der Einnahme von Ertugliflozin in Einzeldosen von bis zu 300 mg sowie Mehrfachdosen von bis zu 100 mg täglich über 2 Wochen. Es wurden keine potentiell akuten Symptome oder Anzeichen einer Überdosierung beobachtet. Die Elimination von Ertugliflozin mittels Hämodialyse wurde nicht untersucht.
Sitagliptin
Im Rahmen kontrollierter klinischer Studien mit gesunden Probanden wurden Einzeldosen von bis zu 800 mg Sitagliptin gegeben. Minimale QTc‑Verlängerungen, die nicht als klinisch relevant erachtet wurden, wurden in einer Studie unter einer Dosis von 800 mg Sitagliptin beobachtet. Es gibt keine Erfahrungen mit Dosen über 800 mg aus klinischen Studien. In Phase I Mehrfachdosisstudien wurden keine dosisabhängigen klinischen Nebenwirkungen unter Dosen von bis zu 600 mg pro Tag über Zeiträume bis zu 10 Tagen und 400 mg pro Tag über Zeiträume bis zu 28 Tagen unter Sitagliptin beobachtet.
Sitagliptin ist in geringem Umfang dialysierbar. In klinischen Studien wurden ca. 13,5 % der Dosis während einer 3 − 4‑stündigen Hämodialyse eliminiert. Eine verlängerte Hämodialyse kann in Betracht gezogen werden, wenn dies klinisch angebracht ist. Es ist nicht bekannt, ob Sitagliptin mittels Peritonealdialyse eliminiert werden kann.
Pharmakotherapeutische Gruppe: Antidiabetika, Kombinationen oraler Antidiabetika, ATC-Code: A10BD24.
Wirkmechanismus
Steglujan kombiniert zwei antidiabetische Wirkstoffe mit komplementären Wirkmechanismen zur Verbesserung der Blutzuckerkontrolle bei Patienten mit Typ‑2 Diabetes: Ertugliflozin, ein SGLT2-Inhibitor und Sitagliptinphosphat, ein DPP‑4-Inhibitor.
Ertugliflozin
SGLT2 ist der Haupttransporter, der für die Rückresorption von Glucose aus dem glomerulären Filtrat in den Kreislauf verantwortlich ist. Ertugliflozin ist ein potenter, selektiver und reversibler Inhibitor von SGLT2. Durch die Hemmung von SGLT2 verringert Ertugliflozin die Rückresorption von renal filtrierter Glucose und senkt die Nierenschwelle für Glucose ab und steigert somit die Glucoseausscheidung im Urin.
Sitagliptin
Sitagliptin gehört zu einer Substanzklasse oraler Antidiabetika, den sog. DPP‑4-Inhibitoren, welche die Blutzuckerkontrolle bei Typ‑2 Diabetikern wahrscheinlich dadurch verbessern, indem sie die Spiegel der aktiven Inkretinhormone anheben. Inkretinhormone, wie das Glucagon-like-Peptide-1 (GLP‑1) und das Glucose-dependent insulinotropic Polypeptide (GIP), werden vom Darm über den Tag hinweg in die Blutbahn freigesetzt, und ihre Spiegel steigen als Reaktion auf eine Mahlzeit an. Die Inkretine sind Teil eines endogenen Systems, das bei der physiologischen Regulation der Glucosehomöostase eine Rolle spielt. Wenn die Blutglucosespiegel normal oder erhöht sind, erhöhen GLP‑1 und GIP die Insulinsynthese und -freisetzung aus den Beta-Zellen des Pankreas über intrazelluläre Signalwege unter Beteiligung von cyclischem Adenosinmonophosphat (cAMP). In Tiermodellen zu Typ‑2 Diabetes zeigte die Behandlung mit GLP‑1 oder mit DPP‑4-Inhibitoren eine Verbesserung der Sensibilität der Beta-Zellen gegenüber Glucose und regte die Insulinsynthese und ‑freisetzung an. Bei höheren Insulinspiegeln wird die Glucoseaufnahme in das Gewebe verstärkt. Zusätzlich senkt GLP‑1 die Glucagonfreisetzung aus den Alpha-Zellen des Pankreas. Verringerte Glucagonkonzentrationen führen zusammen mit erhöhten Insulinspiegeln zu einer verminderten hepatischen Glucoseproduktion. Dies führt zur Senkung der Blutglucosespiegel. Die Wirkungen von GLP‑1 und GIP sind glucoseabhängig, so dass bei niedrigen Blutglucosespiegeln weder eine Stimulation der Insulinfreisetzung noch die Unterdrückung der Glucagonfreisetzung durch GLP‑1 beobachtet werden. Sowohl für GLP‑1 als auch GIP gilt, dass bei Glucoseanstieg über den Normalwert die Insulinfreisetzung verstärkt angeregt wird. GLP‑1 beeinträchtigt zudem die normale Glucagonreaktion auf Hypoglykämien nicht. Die Aktivität von GLP‑1 und GIP wird durch das Enzym DPP‑4 begrenzt, welches die Inkretine rasch zu inaktiven Produkten abbaut. Sitagliptin verhindert den durch DPP‑4 bedingten Abbau der Inkretine und erhöht somit die Plasmakonzentrationen der aktiven Formen von GLP‑1 und GIP. Indem Sitagliptin die Spiegel aktiver Inkretine erhöht, steigert es die Insulinfreisetzung und senkt die Glucagonspiegel jeweils glucoseabhängig. Bei Typ‑2 Diabetikern mit Hyperglykämie führen diese Veränderungen der Insulin- und Glucagonspiegel zu einer Reduzierung des HbA1c und niedrigeren Nüchtern- und postprandialen Blutzuckerwerten. Der glucoseabhängige Wirkmechanismus von Sitagliptin unterscheidet sich von dem der Sulfonylharnstoffe, welche auch bei niedrigen Glucosespiegeln die Insulinfreisetzung erhöhen, was bei Typ‑2 Diabetikern und gesunden Probanden zu Hypoglykämien führen kann. Sitagliptin ist ein potenter und hoch selektiver Inhibitor des DPP‑4 Enzyms und hemmt in therapeutischen Konzentrationen nicht die eng verwandten Enzyme DPP‑8 oder DPP‑9.
In einer zweitägigen Studie mit gesunden Probanden erhöhte Sitagliptin allein die aktiven GLP‑1-Konzentrationen, während Metformin allein die aktiven und Gesamt-GLP‑1-Konzentrationen in ähnlichem Ausmaß erhöhte. Die gemeinsame Anwendung von Sitagliptin und Metformin hatte eine additive Wirkung auf die aktiven GLP‑1-Konzentrationen. Sitagliptin erhöhte die aktiven GIP-Konzentrationen, nicht aber Metformin.
Pharmakodynamische Wirkungen
Ertugliflozin
Glucoseausscheidung im Urin und Urinvolumen
Bei gesunden Probanden und bei Patienten mit Typ‑2 Diabetes mellitus konnte eine dosisabhängige Steigerung der Glucoseausscheidung im Urin nach Einzel- und Mehrfachgabe von Ertugliflozin beobachtet werden. Dosis-Wirkungs-Modelle zeigen, dass 5 mg und 15 mg Ertugliflozin bei Patienten mit Typ‑2 Diabetes mellitus zu einer annähernd maximalen Glucoseausscheidung im Urin führen, was 87 % bzw. 96 % der maximalen Hemmung entspricht.
Klinische Wirksamkeit und Sicherheit
Blutzuckersenkung
Die glykämische Wirksamkeit und Sicherheit von Ertugliflozin in Kombination mit Sitagliptin wurde im Rahmen von 3 multizentrischen, randomisierten, doppelblinden, placebo- und aktivkontrollierten, klinischen Phase III Studien an insgesamt 1 985 Patienten mit Typ‑2 Diabetes untersucht. Über die 3 Studien hinweg waren von den eingeschlossenen Patienten 72,9 % bis 90,4 % kaukasischer, 0 % bis 20,3 % asiatischer, 1,9 % bis 4,5 % schwarzafrikanischer und 4,8 % bis 5,4 % sonstiger ethnischer Herkunft. Patienten mit hispanischer oder lateinamerikanischer Herkunft waren in der Gesamtpopulation zu 15,6 % bis 36,1 % vertreten. Über die 3 Studien hinweg hatten die Patienten ein Durchschnittsalter von 55,1 bis 59,1 Jahren (Spanne 21 Jahre bis 85 Jahre), 16,2 % bis 29,9 % der Patienten waren ≥ 65 Jahre und 2,3 % bis 2,8 % der Patienten waren ≥ 75 Jahre alt.
Faktorielle Studie zur Untersuchung von Ertugliflozin und Sitagliptin als Add-on-Kombinationstherapie mit Metformin
Insgesamt 1 233 Patienten mit Typ‑2 Diabetes wurden im Rahmen einer randomisierten, doppelblinden, multizentrischen, 26‑wöchigen, aktivkontrollierten Studie zur Untersuchung der Wirksamkeit und Sicherheit von Ertugliflozin 5 mg oder 15 mg in Kombination mit 100 mg Sitagliptin im Vergleich zu den jeweiligen Einzelkomponenten eingeschlossen. Die Patienten mit Typ‑2 Diabetes, deren Blutzucker durch eine Metformin-Monotherapie (≥ 1 500 mg/Tag) nicht ausreichend kontrolliert werden konnte, wurden randomisiert einem der fünf aktiven Behandlungsarme zugeteilt: Ertugliflozin 5 mg oder 15 mg, 100 mg Sitagliptin oder 100 mg Sitagliptin in Kombination mit Ertugliflozin 5 mg oder 15 mg, jeweils zur einmal täglichen Einnahme zusätzlich zur Fortsetzung einer Hintergrundtherapie mit Metformin (siehe Tabelle 2).
Tabelle 2: Ergebnisse einer faktoriellen Studie über 26 Wochen zur Untersuchung von Ertugliflozin und Sitagliptin als Add-on-Kombinationstherapie mit Metformin im Vergleich zu den jeweiligen Einzelkomponenten*
Ertugliflozin 5 mg | Ertugliflozin 15 mg | Sitagliptin 100 mg | Ertugliflozin 5 mg | Ertugliflozin 15 mg | ||
HbA1c (%) | N = 250 | N = 248 | N = 247 | N = 243 | N = 244 | |
Ausgangswert | 8,6 | 8,6 | 8,5 | 8,6 | 8,6 | |
Abweichung vom Ausgangswert (LS‑Mittelwert†) | −1,0 | −1,1 | −1,1 | −1,5 | −1,5 | |
Differenz zu | ||||||
Sitagliptin | −0,4‡ (−0,6; −0,3) | −0,5‡ (−0,6; −0,3) | ||||
Ertugliflozin 5 mg | −0,5‡ (−0,6; −0,3) | |||||
Ertugliflozin 15 mg | −0,4‡ (−0,6; −0,3) | |||||
(LS‑Mittelwert†, 95 % KI) | ||||||
Patienten [N (%)] mit HbA1c < 7 % | 66 (26,4) | 79 (31,9) | 81 (32,8) | 127 (52,3)§ | 120 (49,2)§ | |
Körpergewicht (kg) | N = 250 | N = 248 | N = 247 | N = 243 | N = 244 | |
Ausgangswert | 88,6 | 88,0 | 89,8 | 89,5 | 87,5 | |
Abweichung vom Ausgangswert (LS‑Mittelwert†) | −2,7 | −3,7 | −0,7 | −2,5 | −2,9 | |
Differenz zu Sitagliptin (LS‑Mittelwert†, 95 % KI) | −1,8‡ (−2,5; −1,2) | −2,3‡ (−2,9; −1,6) | ||||
* N beinhaltet alle randomisiert behandelten Patienten mit mindestens einer Messung der jeweiligen Zielgröße. | ||||||
Ertugliflozin als Add-on-Kombinationstherapie mit Metformin und Sitagliptin
Insgesamt 463 Patienten mit Typ‑2 Diabetes, deren Blutzucker durch eine Therapie mit Metformin (≥ 1 500 mg/Tag) und einmal täglich 100 mg Sitagliptin nicht ausreichend kontrolliert werden konnte, wurden im Rahmen einer randomisierten, doppelblinden, multizentrischen, 26‑wöchigen, placebokontrollierten Studie zur Untersuchung der Wirksamkeit und Sicherheit von Ertugliflozin eingeschlossen. Die Patienten erhielten zusätzlich zu einer fortgesetzten Hintergrundtherapie mit Metformin und Sitagliptin randomisiert einmal täglich Ertugliflozin 5 mg, Ertugliflozin 15 mg oder Placebo (siehe Tabelle 3).
Tabelle 3: Ergebnisse einer Add-on Studie über 26 Wochen zur Untersuchung von Ertugliflozin in Kombination mit Metformin und Sitagliptin*
Ertugliflozin 5 mg | Ertugliflozin 15 mg | Placebo | |
HbA1c (%) | N = 156 | N = 153 | N = 153 |
Ausgangswert (Mittelwert) | 8,1 | 8,0 | 8,0 |
Abweichung vom Ausgangswert (LS‑Mittelwert†) | −0,8 | −0,9 | −0,1 |
Differenz zu Placebo (LS‑Mittelwert†, 95 % KI) | −0,7‡ (−0,9; −0,5) | −0,8‡ (−0,9; −0,6) | |
Patienten [N (%)] mit HbA1c < 7 % | 50 (32,1)§ | 61 (39,9)§ | 26 (17,0) |
Körpergewicht (kg) | N = 156 | N = 153 | N = 153 |
Ausgangswert (Mittelwert) | 87,6 | 86,6 | 86,5 |
Abweichung vom Ausgangswert (LS‑Mittelwert†) | −3,3 | −3,0 | −1,3 |
Differenz zu Placebo (LS‑Mittelwert†, 95 % KI) | −2,0‡ (−2,6; −1,4) | −1,7‡ (−2,3; −1,1) | |
* N beinhaltet alle randomisiert behandelten Patienten mit mindestens einer Messung der jeweiligen Zielgröße. | |||
Kombinationstherapie mit Ertugliflozin und Sitagliptin
Insgesamt 291 Patienten mit Typ‑2 Diabetes, deren Blutzucker durch Diät und Bewegung nicht ausreichend kontrolliert werden konnte, wurden im Rahmen einer randomisierten, doppelblinden, multizentrischen, placebokontrollierten, 26‑wöchigen Studie zur Untersuchung der Wirksamkeit und Sicherheit von Ertugliflozin in Kombination mit Sitagliptin eingeschlossen. Die Patienten erhielten ohne antidiabetische Hintergrundtherapie randomisiert einmal täglich Ertugliflozin 5 mg oder Ertugliflozin 15 mg in Kombination mit Sitagliptin (100 mg) oder Placebo (siehe Tabelle 4).
Tabelle 4: Ergebnisse einer Studie über 26 Wochen zur Untersuchung von Ertugliflozin und Sitagliptin als Kombinationstherapie*
Ertugliflozin 5 mg | Ertugliflozin 15 mg | Placebo | |
HbA1c (%) | N = 98 | N = 96 | N = 96 |
Ausgangswert (Mittelwert) | 8,9 | 9,0 | 9,0 |
Abweichung vom Ausgangswert (LS‑Mittelwert†) | −1,6 | −1,7 | −0,4 |
Differenz zu Placebo (LS‑Mittelwert†, 95 % KI) | −1,2‡ (−1,5; −0,8) | −1,2‡ (−1,6; −0,9) | |
Patienten [N (%)] mit HbA1c < 7 % | 35 (35,7)§ | 30 (31,3)§ | 8 (8,3) |
Körpergewicht (kg) | N = 98 | N = 96 | N = 97 |
Ausgangswert (Mittelwert) | 90,8 | 91,3 | 95,0 |
Abweichung vom Ausgangswert (LS‑Mittelwert†) | −2,9 | −3,0 | −0,9 |
Differenz zu Placebo (LS‑Mittelwert†, 95 % KI) | −2,0‡ (−3,0; −1,0) | −2,1‡ (−3,1; −1,1) | |
* N beinhaltet alle Patienten, die mindestens eine Dosis der Studienmedikation erhalten haben und mit mindestens einer Messung der jeweiligen Zielgröße. | |||
Nüchternplasmaglucosespiegel
Im Rahmen von drei placebokontrollierten Studien führte die Behandlung mit Ertugliflozin zu einer statistisch signifikanten Senkung der Nüchternplasmaglucosespiegel (fasting plasma glucose, FPG). Die jeweilige Senkung der FPG lag für Ertugliflozin 5 mg und 15 mg in Differenz zu Placebo in der Monotherapie bei 1,92 bzw. 2,44 mmol/l, bei Anwendung als Add-on zu Metformin bei 1,48 bzw. 2,12 mmol/l und bei Anwendung als Add-on zu Metformin und Sitagliptin bei 1,40 mmol/l bzw. 1,74 mmol/l.
Die Behandlung mit Ertugliflozin in Kombination mit Sitagliptin führte im Vergleich zur alleinigen Anwendung von Sitagliptin, Ertugliflozin oder Placebo zu einer signifikant stärkeren Senkung der FPG. Die Behandlung mit Ertugliflozin 5 mg oder 15 mg in Kombination mit Sitagliptin führte im Vergleich zur alleinigen Anwendung von Ertugliflozin zu einer schrittweisen Senkung der FPG um 0,46 mmol/l bis 0,65 mmol/l bzw. im Vergleich zur alleinigen Anwendung von Sitagliptin um 1,02 mmol/l bis 1,28 mmol/l. Die Behandlung mit Ertugliflozin 5 mg oder 15 mg in Kombination mit Sitagliptin führte in Differenz zu Placebo zu einer Senkung der FPG um 2,16 mmol/l bzw. 2,56 mmol/l.
Wirksamkeit bei Patienten mit einem HbA1c‑Ausgangswert ≥ 10 %
Im Rahmen der Studie bei Patienten mit HbA1c‑Ausgangswerten von 7,5 − 11 %, deren Blutzucker durch eine Therapie mit Metformin nicht ausreichend kontrolliert werden konnte, betrug die Senkung der HbA1c‑Werte in der Subgruppe der Patienten mit einem Ausgangswert von ≥ 10 % unter Behandlung mit Ertugliflozin 5 mg oder 15 mg in Kombination mit Sitagliptin 2,35 % bzw. 2,66 %, im Vergleich zu 2,10 %, 1,30 % und 1,82 % unter Behandlung mit den Einzelkomponenten Ertugliflozin 5 mg, Ertugliflozin 15 mg bzw. Sitagliptin.
Postprandiale Plasmaglucosespiegel
Die Behandlung mit Ertugliflozin 5 mg und 15 mg als Monotherapie führte in Differenz zu Placebo zu einer statistisch signifikanten Senkung der postprandialen 2‑Stunden-Plasmaglucosespiegel (PPG) um 3,83 mmol/l bzw. 3,74 mmol/l.
Die Behandlung mit Ertugliflozin 5 mg oder 15 mg in Kombination mit Sitagliptin führte in Differenz zu Placebo zu einer statistisch signifikanten Senkung der postprandialen 2‑Stunden-Plasmaglucosespiegel (PPG) um 3,46 mmol/l bzw. 3,87 mmol/l.
Blutdruck
Die Behandlung mit Ertugliflozin 5 mg oder 15 mg in Kombination mit Sitagliptin 100 mg führte nach 26 Wochen zu einer statistisch signifikanten Senkung des systolischen Blutdrucks im Vergleich zu Sitagliptin allein (−2,8 mmHg und −3,0 mmHg für Ertugliflozin/Sitagliptin 5 mg/100 mg bzw. 15 mg/100 mg) oder im Vergleich zu Placebo (−4,4 mmHg und −6,4 mmHg für Ertugliflozin/Sitagliptin 5 mg/100 mg bzw. 15 mg/100 mg). Zudem führte die Behandlung mit Ertugliflozin 5 mg oder 15 mg als Add-on zu einer Hintergrundtherapie mit Metformin und Sitagliptin zu einer statistisch signifikanten Senkung des systolischen Blutdrucks in Differenz zu Placebo von 2,9 mmHg bzw. 3,9 mmHg.
Subgruppenanalyse
Die Verbesserung der HbA1c‑Werte war bei Patienten mit Typ‑2 Diabetes unter Ertugliflozin in Kombination mit Sitagliptin über verschiedene Subgruppen hinweg, definiert nach Alter, Geschlecht, ethnischer Herkunft und Dauer der Typ‑2 Diabeteserkrankung, vergleichbar.
Kardiovaskuläre Endpunkte
Kardiovaskuläre Endpunkt-Studie zu Ertugliflozin (VERTIS-CV)
Die Wirkung von Ertugliflozin auf das kardiovaskuläre Risiko bei erwachsenen Patienten mit Typ-2 Diabetes mellitus und bestehender atherosklerotischer kardiovaskulärer Erkrankung wurde im Rahmen der VERTIS-CV Studie, einer multizentrischen, multinationalen, randomisierten, doppelblinden, placebokontrollierten, endpunktbezogenen Studie, untersucht. Die Studie verglich das Risiko für das Auftreten von schweren kardiovaskulären Ereignissen (major adverse cardiovascular event, MACE) unter Ertugliflozin im Vergleich zu Placebo als Add-on zu Standardtherapien zur Behandlung von Diabetes und atherosklerotischen kardiovaskulären Erkrankungen.
Insgesamt wurden 8 246 Patienten randomisiert (Placebo N = 2 747, Ertugliflozin 5 mg N = 2 752, Ertugliflozin 15 mg N = 2 747) und im Median über 3 Jahre beobachtet. Das mittlere Alter lag bei 64 Jahren und ca. 70 % waren Männer.
Alle in der Studie eingeschlossenen Patienten hatten zu Studienbeginn einen unzureichend kontrollierten Typ-2 Diabetes mellitus (HbA1c ≥ 7 %). Die mittlere Dauer der Typ-2 Diabetes mellitus Erkrankung lag bei 13 Jahren, der mittlere HbA1c-Ausgangswert betrug 8,2 % und die mittlere eGFR lag bei 76 ml/min/1,73 m2. Zu Studienbeginn wurden die Patienten mit einem (32 %) oder mehreren (67 %) blutzuckersenkenden Arzneimitteln einschließlich Metformin (76 %), Insulin (47 %), Sulfonylharnstoff (41 %), DPP‑4 Inhibitoren (11 %) und GLP‑1 Rezeptoragonisten (3 %) behandelt.
Nahezu alle Patienten (99 %) hatten zu Studienbeginn eine bestehende atherosklerotische kardiovaskuläre Erkrankung. Ca. 24 % der Patienten hatten eine Herzinsuffizienz in der Vorgeschichte. Der primäre Endpunkt der VERTIS-CV Studie war die Zeit bis zum erstmaligen Auftreten eines MACE (kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt [MI] oder nicht-tödlicher Schlaganfall).
Ertugliflozin zeigte Nichtunterlegenheit im Vergleich zu Placebo hinsichtlich MACE (siehe Tabelle 5). Die Ergebnisse der Gruppen unter 5 mg und 15 mg Ertugliflozin waren konsistent mit den Ergebnissen der kombinierten Dosis-Gruppen.
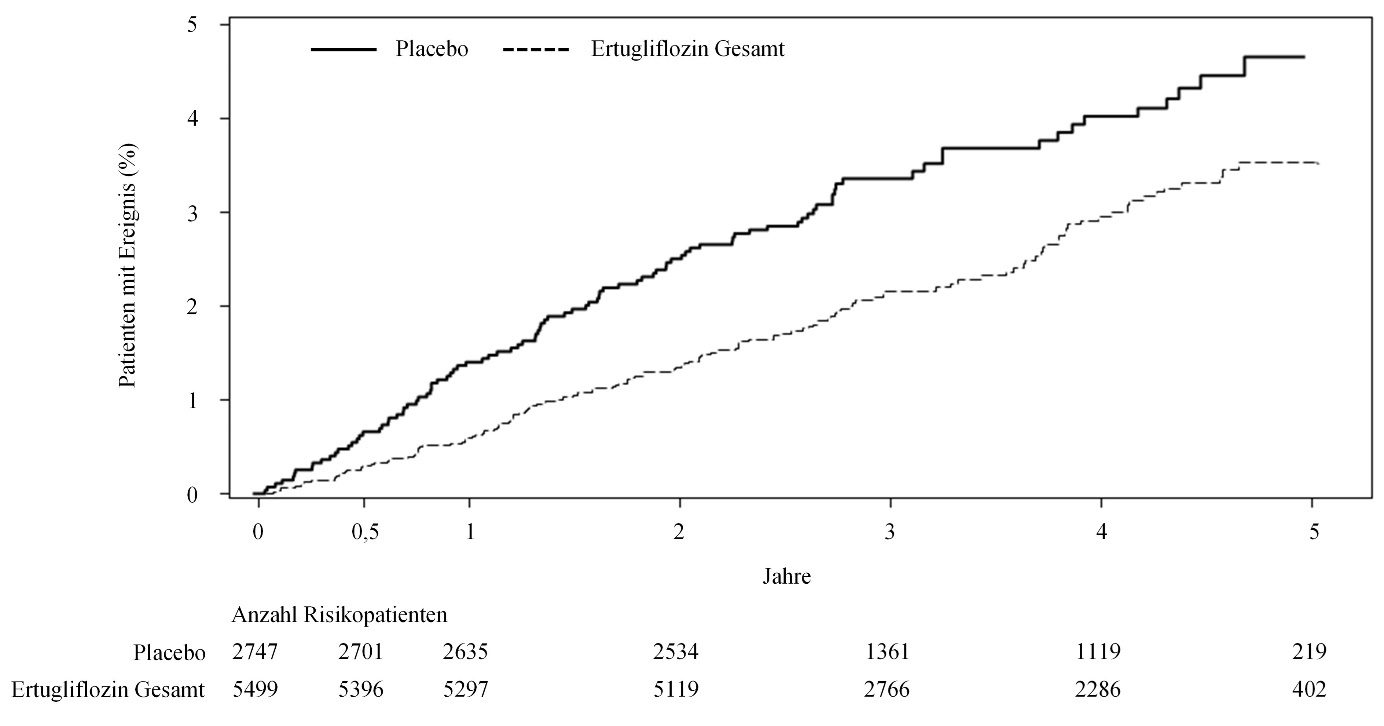
Bei Patienten, die mit Ertugliflozin behandelt wurden, war die Hospitalisierungsrate aufgrund einer Herzinsuffizienz geringer als bei Patienten, die mit Placebo behandelt wurden (siehe Tabelle 5 und Abbildung 1).
Tabelle 5: Auswertung der VERTIS-CV Studie bezüglich MACE, deren Komponenten und Hospitalisierung aufgrund einer Herzinsuffizienz*
Placebo (N = 2 747) | Ertugliflozin (N = 5 499) | ||||
Endpunkt† | N (%) | Ereignisrate (pro 100 Patientenjahre) | N (%) | Ereignisrate (pro 100 Patientenjahre) | Hazard Ratio vs. Placebo |
MACE (CV Tod, nicht-tödlicher MI oder nicht-tödlicher Schlaganfall) | 327 (11,9) | 4,0 | 653 (11,9) | 3,9 | 0,97 |
Nicht-tödlicher MI | 148 (5,4) | 1,6 | 310 (5,6) | 1,7 | 1,04 |
Nicht-tödlicher Schlaganfall | 78 (2,8) | 0,8 | 157 (2,9) | 0,8 | 1,00 |
CV Tod | 184 (6,7) | 1,9 | 341 (6,2) | 1,8 | 0,92 |
Hospitalisierung aufgrund einer Herzinsuffizienz# | 99 (3,6) | 1,1 | 139 (2,5) | 0,7 | 0,70 |
N = Anzahl der Patienten, KI = Konfidenzintervall, CV = kardiovaskulär, MI = Myokardinfarkt. | |||||
Abbildung 1: Zeit bis zum erstmaligen Auftreten des Ereignisses Hospitalisierung aufgrund einer Herzinsuffizienz
Kardiovaskuläre Endpunkt-Studie zu Sitagliptin (TECOS)
Die TECOS-Studie war eine randomisierte Studie mit 14 671 Patienten in der „Intention to Treat Population“ mit einem HbA1c‑Wert von ≥ 6,5 bis 8,0 % und manifester kardiovaskulärer Erkrankung, die zusätzlich zur Standardversorgung, die hinsichtlich HbA1c-Zielwert und kardiovaskulärer Risikofaktoren den lokalen Therapierichtlinien angepasst war, entweder mit Sitagliptin 100 mg pro Tag (7 332 Patienten) (oder 50 mg pro Tag, falls die geschätzte glomeruläre Filtrationsrate [eGFR] zu Studienbeginn bei ≥ 30 und < 50 ml/min/1,73 m2 lag) oder mit Placebo (7 339 Patienten) behandelt wurden. Patienten mit einer eGFR von < 30 ml/min/1,73 m2 wurden nicht in die Studie eingeschlossen. Die Studienpopulation schloss 2 004 Patienten ≥ 75 Jahre und 3 324 Patienten mit eingeschränkter Nierenfunktion (eGFR < 60 ml/min/1,73 m2) ein.
Im Verlauf der Studie betrug die geschätzte mittlere Gesamtdifferenz (SD, Standardabweichung) der HbA1c‑Werte zwischen der Sitagliptin- und der Placebogruppe 0,29 % (0,01), 95 % KI (−0,32; −0,27); p < 0,001. Der primäre kardiovaskuläre Endpunkt setzte sich zusammen aus erstmaligem Auftreten von kardiovaskulärem Tod, nicht-tödlichem Myokardinfarkt, nicht-tödlichem Schlaganfall oder Krankenhauseinweisung aufgrund instabiler Angina pectoris. Sekundäre kardiovaskuläre Endpunkte umfassten das erstmalige Auftreten von kardiovaskulärem Tod, nicht-tödlichem Myokardinfarkt oder nicht-tödlichem Schlaganfall; das erstmalige Auftreten einer der Einzelkomponenten des primären kombinierten Endpunktes; Mortalität jeglicher Ursache und Krankenhauseinweisung aufgrund von Herzinsuffizienz.
Nach einer medianen Nachbeobachtung von 3 Jahren zeigte sich, dass bei Patienten mit Typ‑2 Diabetes die Gabe von Sitagliptin zusätzlich zur üblichen Standardversorgung das Risiko schwerer (major) kardiovaskulärer Ereignisse oder das Risiko einer Krankenhauseinweisung aufgrund von Herzinsuffizienz im Vergleich zur üblichen Standardversorgung ohne Gabe von Sitagliptin nicht erhöhte (siehe Tabelle 6).
Tabelle 6: Inzidenzraten der kombinierten kardiovaskulären Endpunkte sowie wesentlicher sekundärer Endpunkte
Sitagliptin 100 mg | Placebo | |||||
N (%) | Inzidenzrate pro 100 Patientenjahre* | N (%) | Inzidenzrate pro 100 Patientenjahre* | Hazard Ratio | p‑Wert† | |
Analyse der Intention to Treat Population | ||||||
Anzahl der Patienten | 7.332 | 7.339 | ||||
Primärer kombinierter Endpunkt | 839 | 4,1 | 851 | 4,2 | 0,98 | < 0,001 |
Sekundärer kombinierter Endpunkt | 745 | 3,6 | 746 | 3,6 | 0,99 | < 0,001 |
Sekundärer Endpunkt | ||||||
Kardiovaskulärer Tod | 380 | 1,7 | 366 | 1,7 | 1,03 | 0,711 |
Myokardinfarkt gesamt (tödlich und nicht-tödlich) | 300 | 1,4 | 316 | 1,5 | 0,95 | 0,487 |
Schlaganfall gesamt (tödlich und nicht-tödlich) | 178 | 0,8 | 183 | 0,9 | 0,97 | 0,760 |
Krankenhauseinweisung aufgrund instabiler Angina pectoris | 116 | 0,5 | 129 | 0,6 | 0,90 | 0,419 |
Tod jeglicher Ursache | 547 | 2,5 | 537 | 2,5 | 1,01 | 0,875 |
Krankenhauseinweisung aufgrund von Herzinsuffizienz‡ | 228 | 1,1 | 229 | 1,1 | 1,00 | 0,983 |
* Die Inzidenzrate pro 100 Patientenjahre wird berechnet als 100 × (Gesamtanzahl der Patienten mit ≥ 1 Ereignis innerhalb des in Frage kommenden Expositionszeitraums pro Gesamt-Patientenjahre des Nachbeobachtungszeitraums). | ||||||
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Steglujan eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei Typ‑2 Diabetes mellitus gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Steglujan
Für Steglujan konnte Bioäquivalenz im Vergleich zur gemeinsamen Anwendung der entsprechenden Dosen von Ertugliflozin- und Sitagliptin-Tabletten nachgewiesen werden.
Die Auswirkungen einer fettreichen Mahlzeit auf die Pharmakokinetik von Ertugliflozin und Sitagliptin sind hinsichtlich der Gabe als Steglujan oder der Gabe der jeweiligen einzelnen Tabletten vergleichbar. Die Anwendung von Steglujan zu den Mahlzeiten senkte die Cmax von Ertugliflozin um 29 % und hatte keine relevante Auswirkung auf die AUCinf von Ertugliflozin oder die AUCinf und Cmax von Sitagliptin.
Ertugliflozin
Allgemeine Einführung
Die Pharmakokinetik von Ertugliflozin ist bei gesunden Probanden und Patienten mit Typ‑2 Diabetes vergleichbar. Die mittlere Steady State-Plasma-AUC und Cmax lagen bei einmal täglicher Behandlung mit Ertugliflozin 5 mg bei 398 ng·h/ml bzw. 81 ng/ml und bei einmal täglicher Behandlung mit Ertugliflozin 15 mg bei 1 193 ng·h/ml bzw. 268 ng/ml. Der Steady State wird bei einmal täglicher Gabe von Ertugliflozin nach 4 bis 6 Tagen erreicht. Ertugliflozin zeigt keine zeitabhängige Pharmakokinetik und akkumuliert im Plasma bis zu 10 − 40 % nach Mehrfachgabe.
Resorption
Maximale Plasmaspiegel (mediane Zeit bis zur maximalen Plasmakonzentration [Tmax]) treten nüchtern 1 Stunde nach einmaliger oraler Gabe von 5 mg und 15 mg Ertugliflozin auf. Plasma Cmax und AUC steigen dosisproportional für Ertugliflozin bei einmaliger Gabe im Bereich von 0,5 mg bis 300 mg und bei Mehrfachgabe im Bereich von 1 mg bis 100 mg. Die absolute orale Bioverfügbarkeit von Ertugliflozin nach Gabe von 15 mg liegt bei annähernd 100 %.
Die Einnahme von Ertugliflozin zu einer fettreichen und kalorienreichen Mahlzeit senkt die Cmax von Ertugliflozin um 29 % und verlängert die Tmax von Ertugliflozin um 1 Stunde. Die AUC bleibt jedoch im Vergleich zum Nüchternzustand unverändert. Der beobachtete Einfluss von Mahlzeiten auf die Pharmakokinetik von Ertugliflozin wird als klinisch nicht relevant erachtet, so dass Ertugliflozin unabhängig von den Mahlzeiten eingenommen werden kann. Im Rahmen der klinischen Phase III Studien wurde Ertugliflozin unabhängig von den Mahlzeiten eingenommen.
Ertugliflozin ist ein Substrat des P‑Glykoproteins (P‑gp) und des Brustkrebs-Resistenz-Protein (BCRP) Transporters.
Verteilung
Das mittlere Verteilungsvolumen im Steady State für Ertugliflozin liegt nach intravenöser Gabe bei 86 Liter. Die Plasmaproteinbindung von Ertugliflozin liegt bei 93,6 % und ist unabhängig von der Ertugliflozin-Plasmakonzentration. Die Plasmaproteinbindung ist bei Patienten mit eingeschränkter Nieren- oder Leberfunktion nicht nennenswert verändert. Das Blut-Plasma-Konzentrationsverhältnis von Ertugliflozin liegt bei 0,66.
Ertugliflozin ist kein Substrat von organischen Anionen-Transportern (OAT1, OAT3), organischen Kationen-Transportern (OCT1, OCT2) oder organischen Anionen-transportierenden Polypeptiden (OATP1B1, OATP1B3) in vitro.
Biotransformation
Ertugliflozin wird primär mittels Metabolisierung ausgeschieden. Ertugliflozin wird hauptsächlich durch UGT1A9- und UGT2B7-vermittelte O‑Glucuronidierung verstoffwechselt. Die zwei dabei entstehenden Glucuronide sind bei klinisch relevanten Konzentrationen pharmakologisch inaktiv. Die CYP-vermittelte (oxidative) Verstoffwechselung von Ertugliflozin ist minimal (12 %).
Elimination
Die mittlere systemische Plasma-Clearance nach intravenöser Gabe von 100 µg lag bei 11 Liter/h. Die mittlere Eliminationshalbwertszeit bei Typ‑2 Diabetikern mit normaler Nierenfunktion wurde auf Basis einer pharmakokinetischen Populationsanalyse ermittelt und beträgt 17 Stunden. Bei gesunden Probanden wurden nach der oralen Gabe einer [14C]‑Ertugliflozin Lösung ca. 41 % und 50 % der wirkstoffbezogenen Radioaktivität über die Fäzes bzw. Urin ausgeschieden. Nur 1,5 % der gegebenen Dosis wurden unverändert als Ertugliflozin über den Urin und 34 % unverändert als Ertugliflozin über die Fäzes ausgeschieden. Dies ist wahrscheinlich auf die biliäre Exkretion der Glucuronidmetaboliten mit nachfolgender Hydrolyse zum ursprünglichen Wirkstoff zurückzuführen.
Besondere Patientengruppen
Eingeschränkte Nierenfunktion
Im Rahmen einer Phase I Studie zur Untersuchung der klinischen Pharmakologie bei Patienten mit Typ‑2 Diabetes und leichter, moderater oder schwerer Einschränkung der Nierenfunktion (mittels eGFR bestimmt) betrug nach Einmalgabe von 15 mg Ertugliflozin der mittlere Anstieg der AUC von Ertugliflozin das ≤ 1,7‑Fache im Vergleich zu Patienten mit normaler Nierenfunktion. Dieser Anstieg der AUC von Ertugliflozin wird als klinisch nicht relevant erachtet. Zwischen den Patientengruppen mit unterschiedlicher Nierenfunktion gab es keine klinisch relevanten Unterschiede bzgl. der Cmax von Ertugliflozin. Die Glucoseausscheidung im Urin über 24 Stunden nahm mit zunehmendem Schweregrad der Einschränkung der Nierenfunktion ab (siehe Abschnitt 4.4). Die Plasmaproteinbindung von Ertugliflozin war bei Patienten mit eingeschränkter Nierenfunktion unverändert.
Eingeschränkte Leberfunktion
Eine moderate Einschränkung der Leberfunktion (gemäß Child-Pugh-Klassifikation) führte zu keinem Anstieg der Exposition von Ertugliflozin. Die AUC und Cmax von Ertugliflozin nahm um ca. 13 % bzw. ca. 21 % im Vergleich zu Patienten mit normaler Leberfunktion ab. Diese Abnahme der Exposition von Ertugliflozin wird als klinisch nicht relevant erachtet. Es liegen keine klinischen Erfahrungen bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse C) vor. Die Plasmaproteinbindung von Ertugliflozin war bei Patienten mit moderater Einschränkung der Leberfunktion unverändert.
Kinder und Jugendliche
Es wurden keine Studien mit Ertugliflozin bei Kindern und Jugendlichen durchgeführt.
Auswirkungen von Alter, Körpergewicht, Geschlecht und ethnischer Herkunft
Basierend auf den Ergebnissen einer pharmakokinetischen Populationsanalyse haben Alter, Körpergewicht, Geschlecht und ethnische Herkunft keine klinisch relevanten Auswirkungen auf die Pharmakokinetik von Ertugliflozin.
Sitagliptin
Resorption
Nach oraler Gabe einer 100‑mg-Dosis an gesunde Probanden wurde Sitagliptin schnell resorbiert, wobei die mediane Tmax 1 − 4 Stunden nach Einnahme der Dosis auftraten. Die mittlere Plasma-AUC von Sitagliptin betrug 8,52 µM·h, die Cmax 950 nM. Die absolute Bioverfügbarkeit von Sitagliptin beträgt ca. 87 %. Da die Einnahme von Sitagliptin zu einer fettreichen Mahlzeit keinen Einfluss auf die Pharmakokinetik von Sitagliptin hatte, kann Steglujan unabhängig von den Mahlzeiten eingenommen werden.
Die Plasma-AUC von Sitagliptin stieg dosisproportional an. Für die Cmax und die C24 h wurde keine Dosisproportionalität festgestellt (die Cmax stieg mehr, die C24 h weniger als dosisproportional an).
Verteilung
Das mittlere Verteilungsvolumen im Steady State nach intravenöser Gabe einer Einzeldosis von 100 mg Sitagliptin an gesunde Probanden beträgt ca. 198 Liter. Der Anteil reversibel an Plasmaproteine gebundenen Sitagliptins ist gering (38 %).
Biotransformation
Sitagliptin wird vorwiegend unverändert über den Urin eliminiert, seine Metabolisierung spielt eine untergeordnete Rolle. Ca. 79 % von Sitagliptin werden unverändert über den Urin ausgeschieden.
Nach oraler Gabe einer [14C]markierten Sitagliptin-Dosis wurden ca. 16 % der Radioaktivität in Form von Sitagliptin-Metaboliten ausgeschieden. Sechs Metaboliten wurden in Spuren gefunden, die jedoch nicht zu der DPP‑4-inhibitorischen Aktivität von Sitagliptin im Plasma beitragen dürften. In vitro Studien deuten darauf hin, dass CYP3A4, mit Beteiligung von CYP2C8, das hauptverantwortliche Enzym für die begrenzte Metabolisierung von Sitagliptin ist.
In vitro Daten zeigten, dass Sitagliptin kein Inhibitor der CYP-Isoenzyme CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 oder 2B6 und kein Induktor von CYP3A4 und CYP1A2 ist.
Elimination
Innerhalb einer Woche nach oraler Gabe einer [14C]markierten Sitagliptin-Dosis an gesunde Probanden wurden ca. 100 % der gegebenen Radioaktivität über die Fäzes (13 %) oder über den Urin (87 %) ausgeschieden. Die Halbwertzeit (t½) einer oralen 100‑mg-Dosis Sitagliptin betrug ca. 12,4 Stunden. Sitagliptin akkumuliert kaum bei wiederholter Gabe. Die renale Clearance betrug ca. 350 ml/min.
Sitagliptin wird überwiegend renal mit Hilfe aktiver tubulärer Sekretion ausgeschieden. Sitagliptin ist beim Menschen ein Substrat für den organischen Anionentransporter 3 (human organic anionic transporter-3, hOAT3), der an der renalen Elimination von Sitagliptin beteiligt sein könnte. Die klinische Bedeutung von hOAT3 für den Sitagliptin-Transport ist noch unbekannt. Sitagliptin ist auch ein Substrat des P‑Glykoproteins (P‑gp), welches ebenfalls an der Vermittlung der renalen Elimination von Sitagliptin beteiligt sein könnte. Jedoch verminderte Ciclosporin, ein P‑gp-Inhibitor, die renale Clearance von Sitagliptin nicht. Sitagliptin ist kein Substrat für den OCT2, OAT1 oder PEPT1/2 vermittelten Transport. Sitagliptin hemmte in vitro bei therapeutisch relevanten Plasmakonzentrationen weder den OAT3 (IC50 = 160 µM), noch den P‑gp (bis zu 250 µM) vermittelten Transport. In einer klinischen Studie hatte Sitagliptin eine geringe Wirkung auf die Plasmakonzentrationen von Digoxin, was darauf hinweist, dass Sitagliptin P‑gp leicht hemmen könnte.
Arzneimittelwechselwirkungen
Es wurden keine Arzneimittelwechselwirkungsstudien mit Steglujan und anderen Arzneimitteln durchgeführt. Diese Studien wurden jedoch mit den Einzelwirkstoffen durchgeführt.
In vitro Untersuchungen von Ertugliflozin
Im Rahmen von in vitro Studien zeigten Ertugliflozin und Ertugliflozin-Glucuronide weder eine Hemmung oder Inaktivierung der CYP450 Isoenzyme CYP1A2, 2C9, 2C19, 2C8, 2B6, 2D6 oder 3A4 noch eine Induktion von CYP1A2, 2B6 oder 3A4. Ertugliflozin und Ertugliflozin-Glucuronide zeigten keine Hemmung der Aktivität von UGT1A6, 1A9 oder 2B7 in vitro. Ertugliflozin zeigte in höheren, klinisch nicht relevanten Konzentrationen eine leichte Hemmung von UGT1A1 und 1A4 in vitro. Ertugliflozin-Glucuronide hatten keinen Einfluss auf diese Isoformen. Insgesamt ist es unwahrscheinlich, dass Ertugliflozin die Pharmakokinetik von gemeinsam angewendeten Arzneimitteln, welche über diese Enzyme eliminiert werden, beeinflusst.
Ertugliflozin oder Ertugliflozin-Glucuronide zeigen in klinisch relevanten Konzentrationen keine relevante Hemmung von P‑gp, OCT2-, OAT1- oder OAT3-Transportern oder von transportierenden Polypeptiden OATP1B1 und OATP1B3 in vitro. Insgesamt ist es unwahrscheinlich, dass Ertugliflozin die Pharmakokinetik von gemeinsam angewendeten Arzneimitteln, welche Substrate dieser Transporter sind, beeinflusst.
In vitro Untersuchungen von Sitagliptin
In vitro Daten legen nahe, dass Sitagliptin CYP450-Isoenzyme weder hemmt noch induziert. Sitagliptin hatte in klinischen Studien keinen relevanten Einfluss auf die Pharmakokinetik von Metformin, Glibenclamid, Simvastatin, Rosiglitazon, Warfarin oder oralen Kontrazeptiva, was in vivo zeigt, dass Sitagliptin eine geringe Neigung zu Wechselwirkungen mit Substraten von CYP3A4, CYP2C8, CYP2C9 und organischen Kationentransportern (OCT) hat. Sitagliptin könnte in vivo ein schwacher Inhibitor des P‑Glykoproteins sein.
In vitro Transportstudien zeigten, dass Sitagliptin ein Substrat für das P‑Glykoprotein und den organischen Anionentransporter 3 (OAT3) ist. Der Transport von Sitagliptin über OAT3 wurde in vitro durch Probenecid gehemmt, wobei jedoch das Risiko klinisch relevanter Wechselwirkungen als gering eingeschätzt wird. Die gleichzeitige Anwendung mit OAT3-Inhibitoren wurde bisher nicht in vivo untersucht.
Besondere Patientengruppen
Die Pharmakokinetik von Sitagliptin war bei gesunden Probanden und Typ‑2 Diabetikern im Allgemeinen ähnlich.
Einschränkung der Nierenfunktion
Bei Patienten mit normaler Nierenfunktion spielt die Metabolisierung, einschließlich der über CYP3A4, nur eine geringe Rolle für die Ausscheidung von Sitagliptin. Bei schwerer Einschränkung der Nierenfunktion oder bei ESRD kann die Metabolisierung möglicherweise eine größere Bedeutung für die Elimination von Sitagliptin haben.
Im Vergleich zur Kontrollgruppe gesunder Probanden kam es bei Patienten mit einer GFR ≥ 45 bis < 90 ml/min zu einem moderaten Anstieg der Plasma-AUC von Sitagliptin. Da Anstiege dieser Größenordnung klinisch nicht relevant sind, ist eine Dosisanpassung bei diesen Patienten nicht erforderlich.
Einschränkung der Leberfunktion
Bei Patienten mit leichter bis moderater Einschränkung der Leberfunktion (Child-Pugh-Score ≤ 9) ist keine Dosisanpassung von Sitagliptin notwendig. Es gibt keine klinischen Erfahrungen mit Sitagliptin bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Score > 9). Da Sitagliptin jedoch überwiegend renal eliminiert wird, ist nicht zu erwarten, dass die Pharmakokinetik von Sitagliptin durch eine schwere Einschränkung der Leberfunktion beeinflusst wird.
Ältere Patienten
Eine altersabhängige Dosisanpassung ist nicht erforderlich. In einer pharmakokinetischen Populationsanalyse der Phase-I- und Phase-II-Studiendaten hatte Alter keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Sitagliptin. Bei älteren Personen (65 bis 80 Jahre) waren die Plasmakonzentrationen von Sitagliptin ca. 19 % höher als bei jüngeren Personen.
Kinder und Jugendliche
Es wurden keine Studien mit Sitagliptin bei Kindern und Jugendlichen durchgeführt.
Weitere Patientengruppen
Eine Dosisanpassung aufgrund von Geschlecht, ethnischer Herkunft oder Body Mass Index (BMI) ist nicht erforderlich. Diese Eigenschaften hatten in einer kombinierten Analyse der pharmakokinetischen Phase-I-Studiendaten und einer pharmakokinetischen Populationsanalyse der Phase-I- und Phase-II-Studiendaten keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Sitagliptin.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, akuten Toxizität, Toxizität bei wiederholter Verabreichung, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Ertugliflozin
Toxizität allgemein
Toxizitätsstudien mit wiederholter oraler Verabreichung wurden an Mäusen, Ratten und Hunden über einen Zeitraum von 13, 26 bzw. 39 Wochen durchgeführt. Anzeichen von unerwünschter Toxizität wurden im Allgemeinen bei Expositionen beobachtet, die mindestens 77‑fach höher waren als beim Menschen (AUC) bei einer für den Menschen empfohlenen Maximaldosis (maximum recommended human dose, MRHD) von 15 mg/Tag. Die meisten Fälle von Toxizität entsprachen der Pharmakologie und standen im Zusammenhang mit renalem Glucoseverlust. Dazu zählten vermindertes Körpergewicht und Körperfett, gesteigerte Nahrungsaufnahme, Diarrhö, Dehydratation, erniedrigte Serumglucosespiegel und Anstieg anderer Serumparameter, die auf einen erhöhten Proteinstoffwechsel hindeuten, Gluconeogenese und Elektrolytungleichgewichte und Veränderungen des Urins wie Polyurie, Glucosurie und Hypercalciurie. Mikroskopische Veränderungen bzgl. Glucosurie und/oder Hypercalciurie wurden nur bei Nagetieren beobachtet und schlossen eine Dilatation der renalen Tubuli, Hypertrophie der Zona glomerulosa in den Nebennieren (Ratten) und einer Zunahme der trabekulären Strukturen im Knochen (Ratten) ein. Außer Erbrechen gab es keine Anzeichen unerwünschter Toxizität bei Hunden bei einer 379‑fach höheren, ungebundenen Exposition als beim Menschen (AUC) bei einer MRHD von 15 mg/Tag.
Kanzerogenität
Im Rahmen der 2‑jährigen Studie zur Untersuchung der Kanzerogenität bei Mäusen wurde Ertugliflozin in Dosen von 5, 15 und 40 mg/kg/Tag oral per Sonde verabreicht. Es gab keine Ertugliflozin-bedingten neoplastischen Befunde bei Dosen bis zu 40 mg/kg/Tag (was basierend auf der AUC einer ca. 41‑fach höheren ungebundenen Exposition als beim Menschen bei einer MRHD von 15 mg/Tag entspricht). Im Rahmen der 2‑jährigen Studie zur Untersuchung der Kanzerogenität bei Ratten wurde Ertugliflozin in Dosen von 1,5, 5 und 15 mg/kg/Tag oral per Sonde verabreicht. Bei männlichen Ratten kam es bei einer Dosis von 15 mg/kg/Tag zu Ertugliflozin-bedingten neoplastischen Befunden wie einem Anstieg der Inzidenz benigner Phäochromozytome im Nebennierenmark. Dieser Befund wurde auf eine unzureichende Kohlehydrataufnahme mit einhergehender veränderter Calciumhomöostase zurückgeführt und wurde als nicht relevantes Risiko für den Menschen erachtet. Die maximale nicht wirksame Dosis (no observed effect level, NOEL) für Neoplasien betrug 5 mg/kg/Tag (ca. 16‑fach höhere ungebundene Exposition als beim Menschen bei einer MRHD von 15 mg/Tag).
Mutagenität
Ertugliflozin war sowohl mit als auch ohne metabolische Aktivierung weder in der mikrobiellen Rückmutation, noch in zytogenetischen in vitro Tests (humane Lymphozyten) oder in in vivo Mikronukleustests an Ratten mutagen oder clastogen.
Reproduktionstoxizität
Im Rahmen der Studie zur Untersuchung der Fertilität und embryonalen Entwicklung bei männlichen und weiblichen Ratten, wurde Ertugliflozin in Dosen von 5, 25 und 250 mg/kg/Tag verabreicht. Es wurden keine Auswirkungen auf die Fertilität bei einer Dosis von 250 mg/kg/Tag beobachtet (was basierend auf der AUC einer ca. 386‑fach höheren ungebundenen Exposition als beim Menschen bei einer MRHD von 15 mg/Tag entspricht). Die Entwicklung von Ratten und Kaninchen wurde durch Ertugliflozin nach maternaler Exposition, die basierend auf der AUC 239‑ bzw. 1 069‑fach höher war als die maximale klinische Dosis beim Menschen von 15 mg/Tag, nicht nachteilig beeinflusst. Bei einer für Ratten maternal toxischen Dosis (250 mg/kg/Tag), welche 510‑fach höher als die maximale klinische Dosis von 15 mg/Tag war, wurden eine verringerte fötale Lebensfähigkeit und eine erhöhte Inzidenz von viszeralen Fehlbildungen beobachtet.
Im Rahmen der Studie zur Untersuchung der prä- und postnatalen Entwicklung bei Ratten wurde nach Verabreichung von Ertugliflozin ≥ 100 mg/kg/Tag (was basierend auf der AUC einer ca. 239‑fach höheren Exposition als beim Menschen bei einer maximalen klinischen Dosis von 15 mg/Tag entspricht) von Tag 6 der Trächtigkeit bis zu Tag 21 der Säugezeit ein verringertes postnatales Wachstum und eine verminderte Entwicklung beobachtet. Bei einer Dosis von 250 mg/kg/Tag war die Geschlechtsreife bei beiden Geschlechtern verzögert (was basierend auf der AUC einer ca. 620‑fach höheren Exposition als beim Menschen bei einer MRHD von 15 mg/Tag entspricht).
Bei der Verabreichung von Ertugliflozin an juvenile Ratten postnatal nach 21 bis 90 Tagen, einem Zeitraum, in dem die Entwicklung der Nieren stattfindet und welcher dem späten zweiten und dritten Trimester der Schwangerschaft beim Menschen entspricht, wurden bei einer Exposition, die basierend auf der AUC 13‑fach höher als die maximale klinische Dosis beim Menschen von 15 mg/Tag war, erhöhtes Gewicht der Nieren, Dilatation der Nierenbecken und Nierentubuli und Mineralisation der Nierentubuli beobachtet. Auswirkungen auf die Knochenentwicklung (Verkürzung des Oberschenkelknochens und Erhöhung des trabekulären Knochenanteils im Oberschenkelknochen) sowie verzögerte Geschlechtsreife wurden bei einer Exposition, die basierend auf der AUC 817‑fach höher als beim Menschen bei einer MRHD von 15 mg/Tag war, beobachtet. Die Auswirkungen auf Nieren- und Knochenentwicklung waren innerhalb der einmonatigen Erholungsphase nicht vollständig reversibel.
Sitagliptin
Bei Nagern wurden Zeichen von Leber- und Nierentoxizität beobachtet bei einer systemischen Expositionsdosis, die dem 58‑Fachen der humantherapeutischen Exposition entsprach, wobei die No-Effect-Dosis (Schwellenwert) dem 19‑Fachen der humantherapeutischen Expositionsdosis entsprach. Bei Ratten wurden Fehlbildungen der Schneidezähne bei der 67‑fachen klinischen Expositionsdosis beobachtet. Die No-Effect-Dosis für diese in einer 14‑wöchigen Rattenstudie erhobenen Befunde entsprach dem 58‑Fachen der humantherapeutischen Dosis. Die Relevanz dieser Ergebnisse für den Menschen ist nicht bekannt. Vorübergehende, auf die Behandlung zurückzuführende, physische Anzeichen, die auf neurotoxische Wirkungen hinweisen, darunter Atmung mit offenem Maul, vermehrter Speichelfluss, Emesis mit weißem Schaum, Ataxie, Zittern, verminderte Aktivität und/oder gekrümmte Haltung wurden bei Hunden bei einer Exposition beobachtet, die der ca. 23‑fachen klinischen Expositionsdosis entsprach. Zusätzlich wurde bei systemischer Exposition, deren Dosis der ca. 23‑fachen humantherapeutischen klinischen Expositionsdosis entsprach, histologisch eine sehr geringe bis geringe Degeneration der Skelettmuskulatur festgestellt. Die No-Effect-Dosis für diese Befunde lag beim 6‑Fachen der humantherapeutischen klinischen Expositionsdosis.
Sitagliptin zeigte in präklinischen Studien keine genotoxischen Wirkungen. Sitagliptin war bei Mäusen nicht kanzerogen. Bei Ratten kam es zu einem häufigeren Auftreten von Leberadenomen und ‑karzinomen bei der 58‑fachen humantherapeutischen Expositionsdosis. Da bei Ratten nachweislich eine hepatotoxische Wirkung mit der Induktion hepatischer Neoplasien einhergeht, ist diese erhöhte Inzidenz hepatischer Tumoren bei Ratten vermutlich ein Sekundäreffekt der chronischen hepatischen Toxizität in dieser hohen Dosis. Aufgrund der hohen Sicherheitsspanne (19‑Faches bei der entsprechenden No-Effect-Dosis) wird diesen neoplastischen Veränderungen keine Relevanz beim Menschen zugemessen.
Nach Gabe von Sitagliptin vor und nach der Paarung wurden weder bei männlichen noch bei weiblichen Ratten unerwünschte Wirkungen auf die Fertilität festgestellt.
In einer prä-/postnatalen Entwicklungsstudie bei Ratten kam es unter Sitagliptin nicht zu unerwünschten Wirkungen.
Studien zur Reproduktionstoxizität zeigten bei Expositionsdosen über dem 29‑Fachen der humantherapeutischen Exposition eine geringfügige, behandlungsbedingte Erhöhung der Inzidenz fetaler Missbildungen der Rippen (fehlende, unterentwickelte und gewellte Rippen) bei den Nachkommen der Ratten. Zeichen maternaler Toxizität wurden bei Kaninchen bei Expositionsdosen über dem 29‑Fachen der humantherapeutischen Exposition beobachtet. Aufgrund der hohen Sicherheitsspanne legen diese Ergebnisse kein relevantes Risiko für die menschliche Fortpflanzung nahe. Sitagliptin tritt in erheblichen Mengen in die Muttermilch laktierender Ratten über (Verhältnis Muttermilch/Plasma: 4:1).
Tablettenkern
Mikrokristalline Cellulose (E 460)
Calciumhydrogenphosphat
Croscarmellose-Natrium
Natriumstearylfumarat (Ph. Eur.) (E 487)
Magnesiumstearat (E 470b)
Propylgallat
Filmüberzug
Hypromellose (E 464)
Hyprolose (E 463)
Titandioxid (E 171)
Eisen(III)-oxid (E 172)
Eisen(III)-hydroxid-oxid x H2O (E 172)
Eisen(II,III)-oxid (E 172)
Carnaubawachs (E 903)
Nicht zutreffend.
2 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Aluminium/PVC/PA/Aluminium Blisterpackung.
Packungen zu 14, 28, 30, 84, 90 und 98 Filmtabletten in nicht perforierten Blisterpackungen.
Packungen zu 30 x 1 Filmtabletten in perforierten Einzeldosis-Blisterpackungen.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Merck Sharp & Dohme B.V.
Waarderweg 39
2031 BN Haarlem
Niederlande
Steglujan 5 mg/100 mg Filmtabletten
EU/1/18/1266/001
EU/1/18/1266/002
EU/1/18/1266/003
EU/1/18/1266/004
EU/1/18/1266/005
EU/1/18/1266/006
EU/1/18/1266/013
Steglujan 15 mg/100 mg Filmtabletten
EU/1/18/1266/007
EU/1/18/1266/008
EU/1/18/1266/009
EU/1/18/1266/010
EU/1/18/1266/011
EU/1/18/1266/012
EU/1/18/1266/014
Datum der Erteilung der Zulassung: 23. März 2018
Datum der letzten Verlängerung der Zulassung: 05. Dezember 2022
September 2024
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
Für weitere Informationen zu diesem Präparat wenden Sie sich bitte an die deutsche Vertretung des Zulassungsinhabers:
MSD Sharp & Dohme GmbH
Levelingstr. 4a
81673 München
Tel.: 0800/673 673 673
Fax: 0800/673 673 329
E-Mail: e-mail@msd.de
Steglujan-GPC/Type II WS-2729
RCN: 000027006-DE
FACH-9000278-0011