
Talzenna® 0,1 mg Hartkapseln
Talzenna® 0,25 mg Hartkapseln
Talzenna® 0,35 mg Hartkapseln
Talzenna® 0,5 mg Hartkapseln
Talzenna® 1 mg Hartkapseln
Talzenna 0,1 mg Hartkapseln
Jede Hartkapsel enthält Talazoparibtosilat, entsprechend 0,1 mg Talazoparib.
Talzenna 0,25 mg Hartkapseln
Jede Hartkapsel enthält Talazoparibtosilat, entsprechend 0,25 mg Talazoparib.
Talzenna 0,35 mg Hartkapseln
Jede Hartkapsel enthält Talazoparibtosilat, entsprechend 0,35 mg Talazoparib.
Talzenna 0,5 mg Hartkapseln
Jede Hartkapsel enthält Talazoparibtosilat, entsprechend 0,5 mg Talazoparib.
Talzenna 1 mg Hartkapseln
Jede Hartkapsel enthält Talazoparibtosilat, entsprechend 1 mg Talazoparib.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Hartkapsel (Kapsel).
Talzenna 0,1 mg Hartkapseln
Undurchsichtige, etwa 14 mm × 5 mm große Hartkapsel mit weißer Kappe (mit schwarzem Aufdruck „Pfizer“) und weißem Unterteil (mit schwarzem Aufdruck „TLZ 0.1“).
Talzenna 0,25 mg Hartkapseln
Undurchsichtige, etwa 14 mm × 5 mm große Hartkapsel mit elfenbeinfarbener Kappe (mit schwarzem Aufdruck „Pfizer“) und weißem Unterteil (mit schwarzem Aufdruck „TLZ 0.25“).
Talzenna 0,35 mg Hartkapseln
Undurchsichtige, etwa 14 mm × 5 mm große Hartkapsel mit elfenbeinfarbener Kappe (mit schwarzem Aufdruck „Pfizer“) und elfenbeinfarbenem Unterteil (mit schwarzem Aufdruck „TLZ 0.35“).
Talzenna 0,5 mg Hartkapseln
Undurchsichtige, etwa 14 mm × 5 mm große Hartkapsel mit hellrosafarbener Kappe (mit schwarzem Aufdruck „Pfizer“) und weißem Unterteil (mit schwarzem Aufdruck „TLZ 0.5“).
Talzenna 1 mg Hartkapseln
Undurchsichtige, etwa 14 mm × 5 mm große Hartkapsel mit hellroter Kappe (mit schwarzem Aufdruck „Pfizer“) und weißem Unterteil (mit schwarzem Aufdruck „TLZ 1“).
Mammakarzinom
Talzenna wird als Monotherapie für die Behandlung von erwachsenen Patienten mit BRCA1/2‑Mutationen in der Keimbahn angewendet, die ein HER2‑negatives, lokal fortgeschrittenes oder metastasiertes Mammakarzinom aufweisen. Die Patienten sollten zuvor mit einem Anthrazyklin und/ oder einem Taxan im (neo)adjuvanten, lokal fortgeschrittenen oder metastasierten Setting behandelt worden sein, es sei denn, sie waren für diese Behandlungen nicht geeignet (siehe Abschnitt 5.1). Patienten mit Hormonrezeptor (HR)-positivem Brustkrebs sollten außerdem bereits eine endokrin-basierte Therapie erhalten haben oder für diese als nicht geeignet eingestuft sein.
Prostatakarzinom
Talzenna wird in Kombination mit Enzalutamid zur Behandlung erwachsener Patienten mit metastasiertem kastrationsresistenten Prostatakarzinom (metastatic castration-resistant prostate cancer, mCRPC) angewendet, bei denen eine Chemotherapie klinisch nicht indiziert ist.
Die Behandlung mit Talzenna sollte von einem in der Anwendung von Krebstherapeutika erfahrenen Arzt durchgeführt und überwacht werden.
Auswahl von Patienten
Mammakarzinom
Die Auswahl der Patienten für die Brustkrebsbehandlung mit Talzenna sollte abhängig vom Nachweis einer pathogenen oder vermutlich pathogenen BRCA‑Keimbahnmutation mittels eines validierten Testverfahrens durch ein erfahrenes Labor erfolgen.
Eine genetische Beratung von Patienten mit BRCA‑Mutationen sollte gemäß nationaler Vorschriften angeboten werden.
Prostatakarzinom
Für die Auswahl von mCRPC-Patienten für eine Behandlung mit Talzenna ist kein Tumormutationstest erforderlich.
Dosierung
Talzenna-Monotherapie (Mammakarzinom)
Die empfohlene Dosis beträgt einmal täglich 1 mg Talazoparib. Die Therapie sollte bis zur Progression der Grunderkrankung oder bis zum Auftreten inakzeptabler Toxizität fortgeführt werden.
Talzenna in Kombination mit Enzalutamid (Prostatakarzinom)
Die empfohlene Dosis beträgt 0,5 mg Talazoparib in Kombination mit 160 mg Enzalutamid einmal täglich. Die Therapie sollte bis zur Progression der Grunderkrankung oder bis zum Auftreten inakzeptabler Toxizität fortgeführt werden.
Bei nicht chirurgisch kastrierten Patienten sollte die medikamentöse Kastration mit einem Gonadotropin Releasing-Hormon(GnRH)-Analogon während der Behandlung fortgesetzt werden.
Angaben zur Dosierungsempfehlung entnehmen Sie bitte der vollständigen Fachinformation zu Enzalutamid.
Auslassen einer Dosis
Falls der Patient sich erbricht oder eine Talzenna-Dosis auslässt, sollte keine zusätzliche Dosis eingenommen werden. Die nächste verordnete Dosis sollte zur üblichen Zeit eingenommen werden.
Dosisanpassungen
Für die Kontrolle unerwünschter Arzneimittelwirkungen sollten je nach Schweregrad und klinischem Erscheinungsbild Unterbrechungen der Behandlung oder Dosisreduktionen in Betracht gezogen werden (siehe Tabelle 1). Die empfohlenen Dosisreduktionsstufen für die Monotherapie mit Talazoparib (Mammakarzinom) und für die Anwendung von Talazoparib in Kombination mit Enzalutamid (Prostatakarzinom) sind in Tabelle 2 bzw. Tabelle 3 aufgeführt.
Vor Beginn der Behandlung mit Talazoparib sollte eine Kontrolle des Differentialblutbilds erfolgen, die anschließend jeden Monat und sofern klinisch indiziert wiederholt werden sollte (siehe Tabelle 1 und Abschnitt 4.4).
Tabelle 1. Dosisanpassungen bei Nebenwirkungen
Unterbrechung der Behandlung mit Talzenna bis zum Erreichen folgender Werte | ||
Hämoglobin < 8 g/dl | ≥ 9 g/dl | Wiederaufnahme der Behandlung mit Talzenna mit der nächstniedrigen Dosierung |
Thrombozytenzahl < 50 000/μl | ≥ 75 000/μl | |
Neutrophilenzahl < 1 000/μl | ≥ 1 500/µl | |
Nicht-hämatologische Nebenwirkung des Grads 3 oder 4 | ≤ Grad 1 | Wiederaufnahme der Behandlung mit Talzenna mit der nächstniedrigen Dosierung in Betracht ziehen oder Talzenna endgültig absetzen |
Tabelle 2. Dosisreduktionsstufen für Talazoparib-Monotherapie (Mammakarzinom)
Talazoparib-Dosisstufe (Mammakarzinom) | |
Empfohlene Anfangsdosis | 1 mg einmal täglich |
Erste Dosisreduktion | 0,75 mg einmal täglich |
Zweite Dosisreduktion | 0,5 mg einmal täglich |
Dritte Dosisreduktion | 0,25 mg einmal täglich |
Tabelle 3. Dosisreduktionsstufen für Talazoparib in Kombination mit Enzalutamid (Prostatakarzinom)
Talazoparib-Dosisstufe (Prostatakarzinom) | |
Empfohlene Anfangsdosis | 0,5 mg einmal täglich |
Erste Dosisreduktion | 0,35 mg einmal täglich |
Zweite Dosisreduktion | 0,25 mg einmal täglich |
Dritte Dosisreduktion | 0,1 mg einmal täglich |
Angaben zur Dosisanpassung bei Nebenwirkungen im Zusammenhang mit Enzalutamid entnehmen Sie bitte der vollständigen Fachinformation zu Enzalutamid.
Die 0,1-mg-Kapseln sind zur Unterstützung von Dosisanpassungen vorgesehen und dürfen nicht gegen andere Stärken ausgetauscht werden.
Gleichzeitige Behandlung mit P-Glykoprotein (P‑gp)-Inhibitoren
Talzenna-Monotherapie (Mammakarzinom)
Starke P‑gp‑Inhibitoren können zu einer erhöhten Talazoparib-Exposition führen. Die gleichzeitige Anwendung von starken P-gp-Inhibitoren während der Behandlung mit Talazoparib sollte vermieden werden. Eine gleichzeitige Anwendung sollte erst nach sorgfältiger Abwägung der möglichen Vorteile und Risiken erfolgen. Wenn die gleichzeitige Anwendung mit starken P‑gp‑Inhibitoren nicht vermeidbar ist, sollte die Dosis von Talzenna auf die nächstniedrigere Dosis reduziert werden. Nach dem Absetzen des starken P‑gp‑Inhibitors kann die Dosis von Talzenna (nach 3–5 Halbwertszeiten des P‑gp‑Inhibitors) auf die vor Beginn der Behandlung mit dem starken P‑gp‑Inhibitor verwendete Dosis erhöht werden (siehe Abschnitt 4.5).
Talzenna in Kombination mit Enzalutamid (Prostatakarzinom)
Die Auswirkungen einer gleichzeitigen Verabreichung von P-gp-Inhibitoren auf die Talazoparib-Exposition bei einer Anwendung von Talazoparib in Kombination mit Enzalutamid wurden nicht untersucht. Daher sollte die gleichzeitige Anwendung von P-gp-Inhibitoren während der Behandlung mit Talazoparib vermieden werden (siehe Abschnitt 4.5).
Besondere Patientengruppen
Leberinsuffizienz
Bei Patienten mit leichter Leberinsuffizienz (Gesamtbilirubin ≤ 1 × obere Normgrenze [upper limit of normal, ULN]) und Aspartat-Aminotransferase [AST] > ULN oder Gesamtbilirubin > 1,0 bis 1,5 × ULN und beliebige AST), mittelschwerer Leberinsuffizienz (Gesamtbilirubin > 1,5 bis 3,0 × ULN und beliebige AST) oder schwerer Leberinsuffizienz (Gesamtbilirubin > 3,0 × ULN und beliebige AST) ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Die Anwendung von Talzenna in Kombination mit Enzalutamid wird für Patienten mit schwerer Leberinsuffizienz (Child-Pugh-Klasse C) nicht empfohlen, da Pharmakokinetik und Sicherheit in dieser Patientengruppe nicht ermittelt wurden (siehe Abschnitt 5.2).
Niereninsuffizienz
Mammakarzinom
Bei Patienten mit leichter Niereninsuffizienz (60 ml/min ≤ Kreatinin‑Clearance [CrCl] < 90 ml/min) ist keine Dosisanpassung erforderlich. Bei Patienten mit mittelschwerer Niereninsuffizienz (30 ml/min ≤ CrCl < 60 ml/min) beträgt die empfohlene Anfangsdosis von Talzenna einmal täglich 0,75 mg. Bei Patienten mit schwerer Niereninsuffizienz (15 ml/min ≤ CrCl < 30 ml/min) beträgt die empfohlene Anfangsdosis von Talzenna einmal täglich 0,5 mg. Es liegen keine Untersuchungen zu Talzenna bei Patienten mit einer CrCl < 15 ml/min oder bei Hämodialyse‑Patienten vor (siehe Abschnitt 5.2).
Prostatakarzinom
Bei Patienten mit leichter Niereninsuffizienz (60 ml/min ≤ Kreatinin-Clearance [CrCl] < 90 ml/min) ist keine Dosisanpassung erforderlich. Bei Patienten mit mittelschwerer Niereninsuffizienz (30 ml/min ≤ CrCl < 60 ml/min) beträgt die empfohlene Dosis von Talzenna einmal täglich 0,35 mg in Kombination mit Enzalutamid einmal täglich peroral. Bei Patienten mit schwerer Niereninsuffizienz (15 ml/min ≤ CrCl < 30 ml/min) beträgt die empfohlene Dosis von Talzenna einmal täglich 0,25 mg in Kombination mit Enzalutamid einmal täglich peroral. Es liegen keine Untersuchungen zu Talzenna bei Patienten mit einer CrCl < 15 ml/min oder bei Hämodialyse‑Patienten vor (siehe Abschnitt 5.2).
Ältere Patienten
Bei älteren Patienten (≥ 65 Jahre) ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Talzenna bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Talzenna ist zur oralen Verabreichung indiziert. Um den Kontakt mit dem Kapselinhalt zu vermeiden, sollten die Kapseln im Ganzen geschluckt und nicht geöffnet oder aufgelöst werden. Talzenna kann unabhängig von den Mahlzeiten angewendet werden (siehe Abschnitt 5.2).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Stillen (siehe Abschnitt 4.6).
Myelosuppression
Bei Patienten, die mit Talazoparib behandelt wurden, wurde über Myelosuppression in Form einer Anämie, Leukopenie/ Neutropenie und/ oder Thrombozytopenie berichtet (siehe Abschnitt 4.8). Die Behandlung mit Talazoparib sollte erst begonnen werden, wenn sich die Patienten von hämatologischen Toxizitäten der vorhergehenden Therapie (≤ Grad 1) erholt haben.
Patienten, die mit Talazoparib behandelt werden, sollten routinemäßig auf hämatologische Parameter und Anzeichen und Symptome einer Anämie, Leukopenie/ Neutropenie und/ oder Thrombozytopenie überwacht werden. Falls es zu solchen Ereignissen kommt, wird eine Dosisanpassung (Reduktion oder Unterbrechung) empfohlen (siehe Abschnitt 4.2). Unterstützende Maßnahmen mit oder ohne Blut- und/ oder Thrombozytentransfusionen und/ oder die Gabe Granulozyten‑koloniestimulierender Faktoren können bei Bedarf erfolgen.
Myelodysplastisches Syndrom/ akute myeloische Leukämie
Bei Patienten, die Poly-(Adenosindiphosphat-Ribose-)Polymerase (PARP)-Inhibitoren, einschließlich Talazoparib, erhielten, wurde über myelodysplastisches Syndrom/ akute myeloische Leukämie (MDS/ AML) berichtet. Insgesamt wurde bei < 1 % der Patienten mit soliden Tumoren, die in klinischen Studien mit Talazoparib behandelt wurden, über MDS/ AML berichtet (siehe Abschnitt 4.8). Faktoren, die zur Entwicklung eines MDS oder einer AML beitragen könnten, sind eine vorangegangene platinhaltige Chemotherapie, andere DNA‑schädigende Wirkstoffe oder Strahlentherapie. Zu Beginn der Behandlung und anschließend monatlich während der Behandlung sollte eine Kontrolle des Differentialblutbilds auf hämatologische Toxizitäten erfolgen. Bei Bestätigung eines MDS oder einer AML sollte Talazoparib abgesetzt werden.
Venöse thromboembolische Ereignisse
Bei mCRPC-Patienten wurde unter Talzenna in Kombination mit Enzalutamid eine höhere Inzidenz venöser thromboembolischer Ereignisse beobachtet als bei alleiniger Verabreichung von Enzalutamid. Patienten sollten auf klinische Anzeichen und Symptome von tiefer Venenthrombose und Lungenembolie überwacht und entsprechend medizinisch behandelt werden (siehe Abschnitt 4.8).
Verhütung bei Frauen im gebärfähigen Alter
Talazoparib erwies sich im In‑vitro‑Test auf Chromosomenaberrationen in humanen Lymphozyten im peripheren Blut und im In‑vivo‑Mikrokerntest im Knochenmark von Ratten als klastogen, war im Ames-Test aber nicht mutagen (siehe Abschnitt 5.3), und könnte bei einer Verabreichung an Schwangere dem Fötus schaden. Schwangere sollten über das mögliche Risiko für den Fetus informiert werden (siehe Abschnitt 4.6). Frauen im gebärfähigen Alter sollten während der Behandlung mit Talzenna nicht schwanger werden und zu Beginn der Behandlung nicht schwanger sein. Vor der Behandlung sollte bei allen Frauen im gebärfähigen Alter ein Schwangerschaftstest durchgeführt werden.
Patientinnen müssen während der Behandlung mit Talzenna und für mindestens 7 Monate nach Abschluss der Behandlung eine hochwirksame Verhütungsmethode anwenden. Da die Anwendung hormoneller Verhütung bei Patientinnen mit Brustkrebs nicht empfohlen wird, sollten zwei nicht hormonelle und komplementäre Verhütungsmethoden angewendet werden (siehe Abschnitt 4.6).
Männlichen Patienten mit Partnerinnen im gebärfähigen Alter oder schwangeren Partnerinnen sollte geraten werden, während der Behandlung mit Talzenna und für mindestens 4 Monate nach Einnahme der letzten Dosis ein wirksames Verhütungsmittel zu benutzen (auch nach Vasektomie).
Talazoparib ist ein Substrat der Arzneimitteltransporter P‑gp und BCRP (breast cancer resistance protein) und wird hauptsächlich als unveränderte Verbindung über die Nieren ausgeschieden.
Wirkstoffe mit möglichen Auswirkungen auf die Plasmakonzentrationen von Talazoparib
P-gp-Inhibitoren
Wirkung von Enzalutamid
Bei gleichzeitiger Anwendung mit 160 mg Enzalutamid erhöht sich die Talazoparib-Exposition ungefähr auf das 2-Fache. Bei einer Verabreichung von Talazoparib 0,5 mg täglich in Kombination mit Enzalutamid wird ungefähr eine vergleichbare Konzentration im Steady State (Ctrough) erreicht wie bei Talazoparib 1 mg pro Tag (siehe Abschnitt 5.2). Wenn Talzenna in Kombination mit Enzalutamid verabreicht wird, beträgt die Anfangsdosis von Talzenna 0,5 mg (siehe Abschnitt 4.2). Die Wechselwirkung anderer Dosen als 160 mg Enzalutamid auf Talazoparib wurde nicht quantifiziert.
Die Auswirkungen einer gleichzeitigen Verabreichung anderer P-gp-Inhibitoren auf die Talazoparib-Exposition bei einer Anwendung von Talazoparib in Kombination mit Enzalutamid wurden nicht untersucht. Wenn eine gleichzeitige Anwendung von P-gp-Inhibitoren bei Verabreichung von Talzenna in Kombination mit Enzalutamid nicht vermieden werden kann, sollte der Patient auf möglicherweise verstärkte Nebenwirkungen überwacht werden.
Wirkung anderer P-gp-Inhibitoren
Daten aus einer Studie zu Arzneimittelwechselwirkungen bei Patienten mit fortgeschrittenen soliden Tumoren wiesen darauf hin, dass sich bei einer gleichzeitigen Anwendung mehrerer Tagesdosen eines P‑gp‑Inhibitors, Itraconazol 100 mg zweimal täglich, mit einer Einzeldosis von 0,5 mg Talazoparib die Gesamtexposition (AUCinf) und maximale Konzentration (Cmax) von Talazoparib um etwa 56 % bzw. 40 % erhöhte, im Vergleich zu der Einzeldosis von 0,5 mg Talazoparib allein. In einer Untersuchung der Populationspharmakokinetik (PK) zeigte sich zudem, dass eine gleichzeitige Anwendung starker P-gp-Inhibitoren die Talazoparib-Exposition im Vergleich zu Talazoparib allein um 45 % erhöhte.
Die gleichzeitige Anwendung starker P‑gp‑Inhibitoren (einschließlich, aber nicht beschränkt auf Amiodaron, Carvedilol, Clarithromycin, Cobicistat, Darunavir, Dronedaron, Erythromycin, Indinavir, Itraconazol, Ketoconazol, Lapatinib, Lopinavir, Propafenon, Chinidin, Ranolazin, Ritonavir, Saquinavir, Telaprevir, Tipranavir und Verapamil) sollte vermieden werden. Wenn die gleichzeitige Anwendung eines starken P‑gp‑Inhibitors nicht vermeidbar ist, sollte die Dosis von Talzenna reduziert werden (siehe Abschnitt 4.2).
P-gp-Induktoren
Daten aus einer Studie zu Arzneimittelwechselwirkungen bei Patienten mit fortgeschrittenen soliden Tumoren zeigten, dass eine gleichzeitige Anwendung einer Einzeldosis 1 mg Talazoparib mit mehreren Tagesdosen des P‑gp‑Induktors Rifampin (Verabreichung von 600 mg Rifampin 30 Minuten vor Talazoparib am Tag der Talazoparib-Dosis) die Cmax von Talazoparib um etwa 37 % erhöhte, im Vergleich zu einer Einzeldosis 1 mg Talazoparib allein. Die AUCinf veränderte sich nicht. Unter den in der Studie zu Arzneimittelwechselwirkungen getesteten Bedingungen ist dies wahrscheinlich die gemeinsame Nettowirkung der P‑gp‑Induktion und ‑Inhibition von Rifampin. Bei einer gleichzeitigen Anwendung mit Rifampin ist keine Anpassung der Talazoparib-Dosis erforderlich. Die Auswirkungen anderer P‑gp‑Induktoren auf die Talazoparib-Exposition wurden jedoch nicht untersucht. Andere P‑gp‑Induktoren (einschließlich, aber nicht beschränkt auf Carbamazepin, Phenytoin und Johanniskraut) könnten die Talazoparib-Exposition verringern.
BCRP-Inhibitoren
Die Auswirkung von BCRP-Inhibitoren auf die PK von Talazoparib wurde in vivo nicht untersucht. Eine gleichzeitige Anwendung von Talazoparib mit BCRP‑Inhibitoren könnte die Talazoparib‑Exposition erhöhen. Eine gleichzeitige Anwendung mit starken BCRP‑Inhibitoren (einschließlich, aber nicht beschränkt auf Curcumin and Ciclosporin) sollte vermieden werden. Wenn die gleichzeitige Anwendung starker BCRP‑Inhibitoren nicht vermieden werden kann, sollten die Patienten auf möglicherweise verstärkte Nebenwirkungen überwacht werden.
Wirkung säurereduzierender Arzneimittel
Die populationspharmakokinetische Analyse weist darauf hin, dass eine gleichzeitige Behandlung mit säurereduzierenden Arzneimitteln, einschließlich Protonenpumpenhemmer und Histaminrezeptor‑2‑Antagonisten (H2RA) oder anderer säurereduzierender Arzneimittel, keine signifikanten Auswirkungen auf die Resorption von Talazoparib hat.
Systemische hormonelle Verhütung
Studien zu Arzneimittelwechselwirkungen zwischen Talazoparib und oralen Kontrazeptiva wurden nicht durchgeführt.
Frauen im gebärfähigen Alter/ Verhütung bei Männern und Frauen
Frauen im gebärfähigen Alter sollten während der Behandlung mit Talzenna nicht schwanger werden und zu Beginn der Behandlung nicht schwanger sein. Vor der Behandlung sollte bei allen Frauen im gebärfähigen Alter ein Schwangerschaftstest durchgeführt werden (siehe Abschnitt 4.4).
Frauen im gebärfähigen Alter müssen vor Beginn der Behandlung mit Talazoparib, während der Behandlung und 7 Monate nach dem Ende der Behandlung mit Talazoparib hochwirksame Verhütungsmethoden anwenden (siehe Abschnitt 4.4). Da die Anwendung hormoneller Verhütung bei Patientinnen mit Brustkrebs nicht empfohlen wird, sollten zwei nicht hormonelle und komplementäre Verhütungsmethoden angewendet werden. Männlichen Patienten mit Partnerinnen im gebärfähigen Alter oder schwangeren Partnerinnen sollte geraten werden, während der Behandlung mit Talzenna und für mindestens 4 Monate nach Einnahme der letzten Dosis ein wirksames Verhütungsmittel zu benutzen (selbst nach Vasektomie, siehe Abschnitt 4.4).
Schwangerschaft
Bisher liegen keine Daten zur Anwendung von Talzenna bei Schwangeren vor. Tierexperimentelle Studien belegen eine embryofetale Toxizität (siehe Abschnitt 5.3). Talzenna kann bei Verabreichung an Schwangere dem Fötus schaden. Die Anwendung von Talzenna während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen (siehe Abschnitt 4.4).
Stillzeit
Es ist nicht bekannt, ob Talzenna in die Muttermilch übergeht. Ein Risiko für gestillte Kinder kann nicht ausgeschlossen werden. Während der Behandlung mit Talzenna und für mindestens 1 Monat nach der letzten Dosis ist das Stillen somit kontraindiziert (Abschnitt 4.3).
Fertilität
Es liegen keine Informationen zur Fertilität von Patienten vor. Basierend auf nicht-klinischen Befunden in Hoden (teilweise reversibel) und Eierstock (reversibel) kann Talzenna die Fertilität zeugungsfähiger Männer beeinträchtigen (siehe Abschnitt 5.3).
Talzenna hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Nach der Gabe von Talazoparib können Fatigue/ Asthenie oder Schwindel auftreten.
Bitte beachten Sie bei der Gabe von Talzenna in Kombination mit Enzalutamid auch die vollständige Fachinformation zu Enzalutamid bezüglich der Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Das Gesamtsicherheitsprofil von Talzenna basiert auf gepoolten Daten von 1 088 Patienten, einschließlich 690 Patienten, die Talazoparib als Monotherapie in einer Dosis von 1 mg pro Tag in klinischen Studien zur Behandlung solider Tumoren erhielten, und 398 mCRPC-Patienten, die Talazoparib 0,5 mg in Kombination mit Enzalutamid 160 mg in der TALAPRO-2-Studie erhielten.
Die häufigsten (≥ 20 %) Nebenwirkungen bei Patienten, die in diesen klinischen Studien mit Talazoparib behandelt wurden, waren Anämie (55,6 %), Fatigue (52,5 %), Übelkeit (35,8 %), Neutropenie (30,3 %), Thrombozytopenie (25,2 %) und Appetit vermindert (21,1 %). Die häufigsten (≥ 10 %) Nebenwirkungen des Grads ≥ 3 von Talazoparib waren Anämie (39,2 %), Neutropenie (16,5 %) und Thrombozytopenie (11,1 %).
Dosisanpassungen (Dosisreduktionen oder -unterbrechungen) aufgrund von Nebenwirkungen kamen bei 58,7 % der mit 1 mg Talzenna als Monotherapie behandelten Patienten vor. Die häufigsten Nebenwirkungen, die zu Dosisanpassungen führten, waren Anämie (33,5 %), Neutropenie (11,7 %) und Thrombozytopenie (9,9 %). Ein endgültiges Absetzen aufgrund einer Nebenwirkung kam bei 2,9 % der mit Talzenna behandelten Patienten vor. Die häufigste Nebenwirkung war Anämie (0,6 %). Die mediane Expositionsdauer betrug 5,6 Monate (Spanne: 0,0–70,2).
Dosisunterbrechungen von Talzenna aufgrund von Nebenwirkungen kamen bei 62,1 % der mCRPC-Patienten vor, die Talzenna in Kombination mit Enzalutamid erhielten, wobei die häufigste Nebenwirkung Anämie (44 %) war. Reduktionen der Talzenna-Dosis aufgrund von Nebenwirkungen kamen bei 52,8 % der Patienten vor, wobei die häufigste Nebenwirkung Anämie (43,2 %) war. Ein endgültiges Absetzen von Talzenna aufgrund von Nebenwirkungen kam bei 18,8 % der Patienten vorm wobei die häufigste Nebenwirkung Anämie (8,3 %) war. Die mediane Expositionsdauer gegenüber Talazoparib betrug 86 Wochen (Spanne: 0,29–186,14).
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen aus dem gepoolten Datensatz werden in Tabelle 4 entsprechend ihrer Systemorganklasse und Häufigkeit gelistet. Die Häufigkeitskategorien sind folgendermaßen definiert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10) und gelegentlich (≥ 1/1 000, < 1/100). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 4. Nebenwirkungen basierend auf einem gepoolten Datensatz aus 8 Studien (n = 1 088)
Systemorganklasse | Alle Schweregrade* | Grad 3 | Grad 4 |
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen) | 2 (0,2) | 1 (< 0,1) | 1 (< 0,1) |
Erkrankungen des Blutes und des Lymphsystems | 274 (25,2) 88 (8,1) | 88 (8,1) 37 (3,4) | 33 (3,0) 4 (0,4) |
Stoffwechsel- und Ernährungsstörungen | 230 (21,1) | 11 (1,0) | 0 (0,0) |
Erkrankungen des Nervensystems | 157 (14,4) 68 (6,3) | 4 (0,4) 0 (0,0) | 1 (< 0,1) 0 (0,0) |
Gefäßerkrankungen | 36 (3,3 %) | 23 (2,1 %) | 2 (0,2 %) |
Erkrankungen des Gastrointestinaltrakts | 167 (15,3) 54 (5,0) | 9 (0,8) 0 (0,0) | 0 (0,0) 0 (0,0) |
Erkrankungen der Haut und des Unterhautzellgewebes | 189 (17,4) | n. z. | n. z. |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | 571 (52,5) | 58 (5,3) | n. z. |
Abkürzungen: n = Anzahl Patienten, n. z. = nicht zutreffend. | |||
Beschreibung ausgewählter Nebenwirkungen
Myelosuppression
Mit einer Myelosuppression in Zusammenhang stehende Nebenwirkungen, Anämie, Neutropenie und Thrombozytopenie, wurden bei Patienten, die Talazoparib erhielten, sehr häufig berichtet. Mit einer Myelosuppression in Zusammenhang stehende Ereignisse des Grads 3 und Grads 4 wurden mit einer Häufigkeit von 37,8 % bzw. 1,5 % der Patienten für Anämie, 15,0 % bzw. 1,6 % für Neutropenie und 8,1 % bzw. 3,0 % für Thrombozytopenie berichtet. Todesfälle aufgrund von mit einer Myelosuppression in Zusammenhang stehenden Nebenwirkungen wurden nicht berichtet.
Die häufigsten in Monotherapie-Studien (Population mit 1 mg/Tag) beobachteten und mit einer Myelosuppression in Zusammenhang stehenden unerwünschten Ereignisse, die zu Dosisanpassungen führten, waren Anämie (33,5 %), Neutropenie (11,7 %) und Thrombozytopenie (9,9 %). Diese wurden bei bis zu etwa 30 % der Patienten in der mit 1 mg/Tag Talazoparib behandelten Population berichtet. Die Nebenwirkung, die zu einem endgültigen Abbruch der Behandlung mit dem Prüfmedikament führte und bei 0,6 % der Patienten auftrat, war Anämie.
Bei mCRPC-Patienten, die Talazoparib in Kombination mit Enzalutamid erhielten, führte Anämie bei 44,0 % der Patienten zu einer Unterbrechung der Talazoparib-Dosis, bei 13,6 % war die Neutrophilenzahl erniedrigt und bei 7,8 % trat eine Verminderung der Thrombozytenzahl auf. Insgesamt waren bei 42,5 % der Patienten Bluttransfusionen erforderlich. Die häufigsten, in 39,2 % der Fälle verwendeten Bluttransfusionen waren Erythrozytenkonzentrate. Eine Unterbrechung aufgrund von Anämie, Neutropenie bzw. Thrombozytopenie trat bei 8,3 %, 3,3 % bzw. 0,5 % der Patienten auf.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das aufgeführte nationale Meldesystem anzuzeigen.
Deutschland
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: https://www.bfarm.de
Österreich
Bundesamt für Sicherheit im Gesundheitswesen
Traisengasse 5
1200 WIEN
ÖSTERREICH
Fax: +43 (0) 50 555 36207
Website: https://www.basg.gv.at/
Es liegen begrenzte Erfahrungen zu einer Überdosierung mit Talazoparib vor. Es wurden keine Nebenwirkungen bei einem Patienten berichtet, der versehentlich an Tag 1 dreißig 1‑mg‑Kapseln Talazoparib einnahm und umgehend mit einer Magenspülung behandelt wurde. Die Symptome einer Überdosierung sind nicht bekannt. Im Fall einer Überdosierung sollte die Behandlung mit Talazoparib abgebrochen werden, und die Ärzte sollten eine Magenspülung in Betracht ziehen, allgemeine unterstützende Maßnahmen ergreifen und symptomatisch behandeln.
Pharmakotherapeutische Gruppe: antineoplastische Mittel, andere antineoplastische Mittel, ATC-Code: L01XK04
Wirkmechanismus
Talazoparib ist ein Inhibitor der PARP-Enzyme PARP-1 (IC50 = 0,7 nM) und PARP-2 (IC50 = 0,3 nM). PARP-Enzyme sind an zellulären Signalwegen für die DNA-Schadensantwort beteiligt, z. B. für DNA-Reparatur, Gentranskription und Zelltod. PARP-Inhibitoren (PARPi) wirken sich über 2 Mechanismen zytotoxisch auf Krebszellen aus: durch Inhibition der katalytischen Aktivität der PARP und durch das sogenannte „PARP trapping“, d. h. die Verhinderung der Dissoziation der an einen PARPi gebundenen PARP-Proteine von der DNA-Läsion, wodurch DNA-Reparatur, -Replikation und ‑Transkription verhindert und somit die Apoptose und/ oder der Zelltod verursacht werden. Die Behandlung von Krebszelllinien mit Defekten in DNA-Reparaturgenen mit Talazoparib als Einzelwirkstoff führt zu einer erhöhten Konzentration von γH2AX, einem Marker für DNA‑Doppelstrangbrüche, sowie zu einer verringerten Zellproliferation und erhöhten Apoptose. Die antitumorale Aktivität von Talazoparib wurde auch in einem aus Patientenmaterial abgeleiteten Xenograft (patient-derived xenograft, PDX)‑Brustkrebsmodell mit BRCA‑Mutation nach vorhergehender Patientenbehandlung mit einem platinbasierten Regime sowie in einem Androgenrezeptor(AR)-positiven Prostatakarzinom-Xenograft-Modell beobachtet. In diesen PDX‑Modellen verringerte Talazoparib das Tumorwachstum und erhöhte die γH2AX‑Konzentration und Apoptose im Tumor.
Die antitumorale Wirkung der kombinierten Inhibition der PARP- und AR-Aktivität basiert auf den folgenden Mechanismen: Die Hemmung der AR-Signalübertragung unterdrückt die Expression von Genen für die homologe Rekombinationsreparatur (HRR), einschließlich BRCA1, was zu einer Sensitivität gegenüber einer PARP-Inhibition führt. Es hat sich gezeigt, dass für eine maximale AR‑Funktion eine PARP1-Aktivität erforderlich ist. Somit könnte eine PARP-Inhibition die AR-Signalübertragung verringern und die Sensitivität gegenüber AR-Signalinhibitoren erhöhen. Eine klinische Resistenz gegenüber einer AR-Blockade wird manchmal mit einer Codeletion von RB1 und BRCA2 in Verbindung gebracht, die wiederum mit einer Sensitivität gegenüber einer PARP-Inhibition zusammenhängt.
Kardiale Elektrophysiologie
Der Effekt von Talazoparib auf die Repolarisation des Herzens wurde mithilfe eines zeitangepassten Elektrokardiogramms (EKG) zur Beurteilung der Veränderung des frequenzkorrigierten QT‑Intervalls (QTc) gegenüber der Baseline und der entsprechenden Talazoparib‑Konzentration im Plasma bei 37 Patienten mit fortgeschrittenen soliden Tumoren untersucht. Bei der klinisch empfohlenen Monotherapie-Höchstdosis von 1 mg einmal täglich hatte Talazoparib keinen klinisch relevanten Effekt auf die QTc‑Verlängerung.
Klinische Wirksamkeit und Sicherheit
HER2-negativer, lokal fortgeschrittener oder metastasierter Brustkrebs mit Keimbahnmutation im BRCA-Gen (germline BRCA mutated, gBRCAm)
EMBRACA-Studie
EMBRACA war eine offene, randomisierte, 2-armige, multizentrische Parallelstudie zu Talzenna gegenüber einer Chemotherapie (Capecitabin, Eribulin, Gemcitabin oder Vinorelbin) bei Patienten mit HER2‑negativem, lokal fortgeschrittenen oder metastasierten Brustkrebs mit Keimbahnmutation im BRCA‑Gen, die zuvor höchstens 3 zytotoxische Chemotherapien zur Behandlung der metastasierten oder lokal fortgeschrittenen Erkrankung erhalten hatten. Die Patienten mussten eine Behandlung mit einem Anthrazyklin und/ oder Taxan (außer bei Kontraindikation) in der neoadjuvanten, adjuvanten und/ oder metastasierten Setting erhalten haben. Bei Patienten mit vorhergehender Platintherapie zur Behandlung einer fortgeschrittenen Erkrankung durfte sich während der Platintherapie keine Krankheitsprogression gezeigt haben. Eine vorhergehende Behandlung mit einem PARPi war nicht zulässig.
Von 431 Patienten, die in die Studie EMBRACA randomisiert wurden, wurde bei 408 Patienten (95 %) mittels eines zentral durchgeführten Testverfahrens für klinische Studien eine pathogene oder vermutlich pathogene BRCA‑Keimbahnmutation nachgewiesen. Bei 354 Patienten (82 %) wurde der Verdacht mittels BRACAnalysis CDx bestätigt. Der BRCA‑Mutationsstatus (positiv für breast cancer susceptibility gene 1 [BRCA1] oder breast cancer susceptibility gene 2 [BRCA2]) war in beiden Behandlungsarmen ähnlich.
Insgesamt wurden 431 Patienten im Verhältnis 2:1 auf Talzenna 1-mg-Kapseln einmal täglich oder eine Chemotherapie in Standarddosierung randomisiert. Die Behandlung wurde bis zur Krankheitsprogression oder bis zum Auftreten einer inakzeptablen Toxizität fortgesetzt. Von den 431 in EMBRACA randomisierten Patienten wurden 287 auf den Talzenna‑Arm und 144 auf den Chemotherapie‑Arm randomisiert, stratifiziert nach Anzahl vorangegangener Chemotherapien im metastasierten Erkrankungsstadium (0 versus 1, 2 oder 3), nach Triple-Negativem-Rezeptorstatus (TNBC, triple-negative breast cancer, versus Nicht-TNBC) und nach dem Vorliegen von Metastasen im zentralen Nervensystem (ZNS) in der Vorgeschichte (ja versus nein).
Patientendemografie, Zustand bei der Baseline und Krankheitsmerkmale waren in den Behandlungsarmen insgesamt vergleichbar (siehe Tabelle 5).
Tabelle 5. Demografie, Baseline und Krankheitsmerkmale – EMBRACA-Studie | ||
Talazoparib | Chemotherapie | |
Medianes Alter (Jahre [Spanne]) | 45,0 (27,0; 84,0) | 50,0 (24,0; 88,0) |
Alterskategorie (Jahre), n (%) | ||
< 50 | 182 (63,4 %) | 67 (46,5 %) |
50 bis < 65 | 78 (27,2 %) | 67 (46,5 %) |
≥ 65 | 27 (9,4 %) | 10 (6,9 %) |
Geschlecht, n (%) | ||
Frauen | 283 (98,6 %) | 141 (97,9 %) |
Männer | 4 (1,4 %) | 3 (2,1 %) |
Ethnische Zugehörigkeit, n (%) | ||
Asiaten | 31 (10,8 %) | 16 (11,1 %) |
Schwarze oder Afroamerikaner | 12 (4,2 %) | 1 (0,7 %) |
Kaukasier | 192 (66,9 %) | 108 (75,0 %) |
Andere | 5 (1,7 %) | 1 (0,7 %) |
Keine Angabe | 47 (16,4 %) | 18 (12,5 %) |
ECOG-Performance-Status, n (%) | ||
0 | 153 (53,3 %) | 84 (58,3 %) |
1 | 127 (44,3 %) | 57 (39,6 %) |
2 | 6 (2,1 %) | 2 (1,4 %) |
Keine Angabe | 1 (0,3 %) | 1 (0,7 %) |
Hormonrezeptorstatus, n (%) | ||
HER2-positiv | 0 (0,0 %) | 0 (0,0 %) |
Triple-Negativ | 130 (45,3 %) | 60 (41,7 %) |
Hormonrezeptor‑positiv (ER-positiv oder PgR-positiv) | 157 (54,7 %) | 84 (58,3 %) |
BRCA-Status gemäß Beurteilung in zentralem Labor oder Labor am Prüfzentrum, n (%) | 287 (100,0 %) | 144 (100,0 %) |
Positiv für die Mutation im BRCA1‑Gen | 133 (46,3 %) | 63 (43,8 %) |
Positiv für die Mutation im BRCA2‑Gen | 154 (53,7 %) | 81 (56,3 %) |
Zeit von der ersten Brustkrebsdiagnose bis zur Diagnose einer fortgeschrittenen Brustkrebserkrankung (Jahre) | ||
n | 286 | 144 |
Median | 1,9 | 2,7 |
Minimum, Maximum | 0; 22 | 0; 24 |
Kategorien für die Zeit von der ersten Brustkrebsdiagnose bis zur Diagnose einer fortgeschrittenen Brustkrebserkrankung | ||
< 12 Monate | 108 (37,6 %) | 42 (29,2 %) |
≥ 12 Monate | 178 (62,0 %) | 102 (70,8 %) |
Anzahl vorangegangener zytotoxischer Regimes zur Behandlung einer lokal fortgeschrittenen oder metastasierten Erkrankung | ||
Mittelwert (Standardabweichung) | 0,9 (1,01) | 0,9 (0,89) |
Median | 1 | 1 |
Minimum, Maximum | 0; 4 | 0; 3 |
Anzahl Patienten mit vorangegangenen zytotoxischen Regimes zur Behandlung einer lokal fortgeschrittenen oder metastasierten Erkrankung, n (%) | ||
0 | 111 (38,7 %) | 54 (37,5 %) |
1 | 107 (37,3 %) | 54 (37,5 %) |
2 | 57 (19,9 %) | 28 (19,4 %) |
3 | 11 (3,8 %) | 8 (5,6 %) |
≥ 4 | 1 (0,3 %) | 0 (0,0 %) |
Anzahl Patienten mit folgenden vorangegangenen Therapien, n (%) | ||
Taxan | 262 (91,3 %) | 130 (90,3 %) |
Anthrazyklin | 243 (84,7 %) | 115 (79,9 %) |
Platin | 46 (16,0 %) | 30 (20,8 %) |
Abkürzungen: BRCA = breast cancer susceptibility gene; ER = Östrogen-Rezeptor (estrogen receptor); HER2 = humaner epidermaler Wachstumsfaktor-Rezeptor 2 (human epidermal growth factor receptor 2); n = Anzahl Patienten; PgR = Progesteron-Rezeptor.
Der primäre Wirksamkeitsendpunkt war das progressionsfreie Überleben (progression-free survival, PFS), bewertet nach den Kriterien für die Bewertung des Ansprechens der Behandlung bei soliden Tumoren (Response Evaluation Criteria In Solid Tumors [RECIST], Version 1.1.) gemäß verblindeter unabhängiger zentraler Beurteilung (blinded independent central review, BICR). Die Sekundärziele waren objektive Ansprechrate (objective response rate, ORR), Gesamtüberleben (overall survival, OS), Sicherheit und PK.
Die Studie zeigte eine statistisch signifikante Verbesserung des PFS, des primären Wirksamkeitsergebnisses, für Talzenna im Vergleich zur Chemotherapie. Zum Zeitpunkt der abschließenden Analyse des OS zeigte sich kein statistisch signifikanter Effekt auf das Gesamtüberleben. Die Wirksamkeitsdaten für EMBRACA sind in Tabelle 6 dargestellt. Die Kaplan‑Meier‑Kurven für das PFS sind in Abbildung 1 und für das Gesamtüberleben in Abbildung 3 dargestellt.
Tabelle 6. Zusammenfassung der Wirksamkeitsergebnisse – EMBRACA-Studie* | ||
Talazoparib | Chemotherapie | |
PFS gemäß BICR | n = 287 | n = 144 |
Ereignisse, Anzahl (%) | 186 (65 %) | 83 (58 %) |
Median (95 %-KI), Monate | 8,6 (7,2; 9,3) | 5,6 (4,2; 6,7) |
Hazard-Ratioa (95 %-KI) | 0,54 (0,41; 0,71) | |
Zweiseitiger p-Wertb | p < 0,0001 | |
OS (abschließende Analyse)c | n = 287 | n = 144 |
Ereignisse, Anzahl (%) | 216 (75,3 %) | 108 (75 %) |
Median (95 %-KI), Monate | 19,3 (16,6; 22,5) | 19,5 (17,4; 22,4) |
Hazard-Ratioa (95 %-KI) | 0,85 (0,67; 1,07)c | |
Zweiseitiger p-Wertb | p = 0,1693 | |
Objektives Ansprechen gemäß Prüfarztd,e | n = 219 | n = 114 |
ORR, % (95 %-KI) | 62,6 (55,8; 69,0) | 27,2 (19,3; 36,3) |
Odds-Ratio (95 %-KI) | 4,99 (2,93; 8,83) | |
Zweiseitiger p-Wertf | p < 0,0001 | |
Dauer des Ansprechens gemäß Prüfarztd | n = 137 | n = 31 |
Median (IQR), Monate | 5,4 (2,8; 11,2) | 3,1 (2,4; 6,7) |
Abkürzungen: BICR = verblindete unabhängige zentrale Beurteilung (blinded independent central review); KI = Konfidenzintervall; CMH = Cochran-Mantel‑Haenszel; CR = komplettes Ansprechen (complete response); IQR = Interquartilbereich (interquartile range); ITT = Intent‑to‑Treat; n = Anzahl Patienten; ORR = objektive Ansprechrate (objective response rate); OS = Gesamtüberleben (overall survival); PARP = Poly-(Adenosindiphosphat)-Ribose-Polymerase; PFS = progressionsfreies Überleben (progression-free survival); PR = partielles Ansprechen (partial response); RECIST 1.1 = Response Evaluation Criteria in Solid Tumors (Kriterien für die Bewertung des Ansprechens der Behandlung bei soliden Tumoren) Version 1.1. | ||
Abbildung 1. Kaplan-Meier-Plot des PFS – EMBRACA-Studie
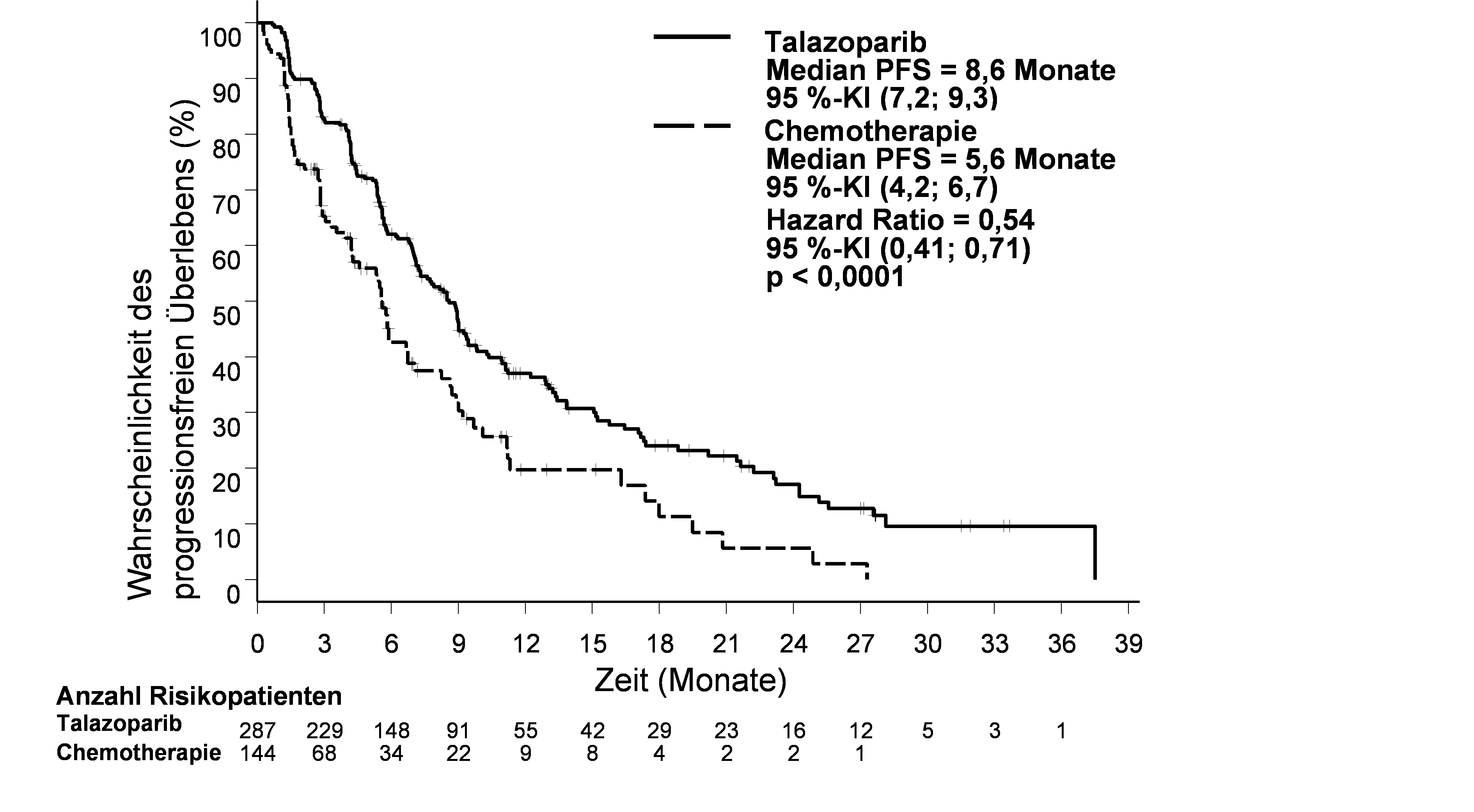
Abkürzungen: KI = Konfidenzintervall; PFS = progressionsfreies Überleben (progression-free survival).
Es wurden mehrere präspezifizierte Subgruppen-PFS-Analysen auf Grundlage der Prognosefaktoren und Baseline-Patientenmerkmalen durchgeführt, um die interne Konsistenz des Behandlungseffekts zu untersuchen. Übereinstimmend mit den Gesamtergebnissen wurde eine Verringerung des Risikos für eine Krankheitsprogression oder Tod zugunsten des Talazoparib‑Arms in allen untersuchten Patienten‑Subgruppen beobachtet (Abbildung 2).
Abbildung 2. Forest-Plot für die PFS-Analysen in den wichtigsten Untergruppen – EMBRACA-Studie
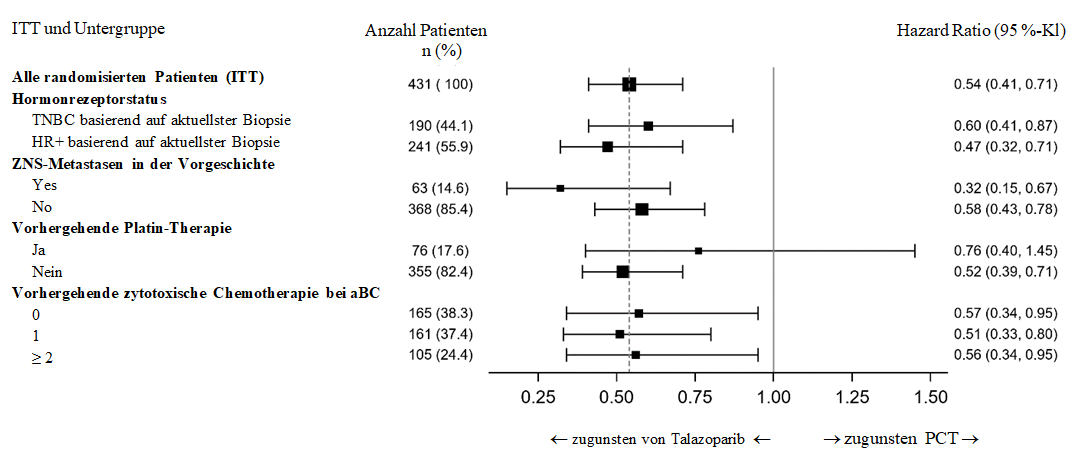
Abkürzungen: aBC = fortgeschrittener Brustkrebs (advanced breast cancer); KI = Konfidenzintervall; ZNS = zentrales Nervensystem; HR+ = Hormonrezeptor-positiv; ITT = Intent-to-Treat; PCT = Behandlung nach Wahl des Arztes, physician’s choice treatment (Chemotherapie); PFS = progressionsfreies Überleben (progression-free survival); TNBC = Triple-Negativer-Brustkrebs (triple-negative breast cancer).
Abbildung 3 Kaplan-Meier-Plot des Gesamtüberlebens – EMBRACA-Studie
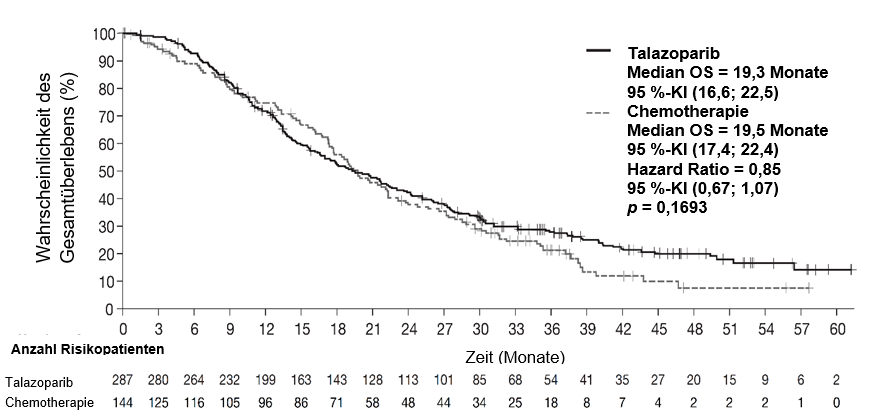
Abkürzungen: KI = Konfidenzintervall; OS = Gesamtüberleben (overall survival).
p-Wert der Primäranalyse basierend auf stratifiziertem Log-rank-Test.
Metastasiertes, kastrationsresistentes Prostatakarzinom (mCRPC)
TALAPRO-2-Studie
TALAPRO-2 war eine randomisierte, doppelblinde, placebokontrollierte Studie, in der Patienten (n = 805) mit mCRPC im Verhältnis 1:1 auf eine Behandlung mit 0,5 mg Talzenna einmal täglich in Kombination mit 160 mg Enzalutamid einmal täglich oder auf einen Vergleichsarm mit Placebo in Kombination mit 160 mg Enzalutamid einmal täglich randomisiert wurden. Alle Patienten erhielten ein Gonadotropin Releasing-Hormon(GnRH)-Analogon oder hatten sich zuvor einer beidseitigen Orchiektomie unterzogen und mussten eine Krankheitsprogression unter einer vorhergehenden Androgendeprivationstherapie aufweisen. Eine vorherige Behandlung mit Abirateron oder eine Chemotherapie auf Taxanbasis bei metastasiertem hormonsensitiven Prostatakarzinom (mHSPC) war zulässig.
Die Randomisierung wurde stratifiziert (1) nach vorheriger Behandlung mit Abirateron oder Taxan-basierter Chemotherapie versus keiner solchen vorhergehenden Behandlung und (2) nach dem mittels prospektiver Next-Generation-Sequenzierung an Tumorgewebe unter Verwendung von FoundationOne CDx oder zirkulierender Tumor-DNA (ctDNA) unter Verwendung von FoundationOne Liquid CDx ermittelten HRR-Genmutationsstatus: Patienten mit HRR-Genmutationen im Tumor (ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, oder RAD51C) versus Patienten mit Tumoren ohne HRR-Genmutationen oder mit unbekanntem Status.
Das mediane Alter betrug in beiden Gruppen 71 Jahre (Spanne 36–91 Jahre), wobei 62 % der Teilnehmer Kaukasier, 31 % Asiaten und 2 % Schwarze waren. Die meisten Teilnehmer (66 %) in beiden Gruppen hatten einen ECOG-Performance-Status von 0. Bei den mit Talzenna behandelten Patienten lag der Anteil der Patienten mit einer nach RECIST 1.1 messbaren Erkrankung bei der Baseline gemäß BICR bei 30 %. Achtundzwanzig Prozent (28 %) der Patienten hatten zuvor Abirateron oder eine Chemotherapie auf Taxanbasis erhalten. Zwanzig Prozent (20 %) hatten Tumoren mit HRR-Genmutationen und 80 % hatten Tumoren ohne HRR-Genmutationen oder mit unbekanntem Status.
Der primäre Wirksamkeitsendpunkt war das radiologische progressionsfreie Überleben (rPFS) gemäß RECIST Version 1.1 und den Kriterien der Prostate Cancer Clinical Trials Working Group Criteria 3 (PCWG3, Knochen) durch ein BICR. Das Gesamtüberleben war ein alpha-kontrollierter sekundärer Endpunkt.
Es wurde eine statistisch signifikante Verbesserung des in der BICR beurteilten rPFS für Talzenna in Kombination mit Enzalutamid im Vergleich zu Placebo in Kombination mit Enzalutamid nachgewiesen. Eine Sensitivitätsanalyse des durch den Prüfarzt beurteilten rPFS stimmte mit den in der BICR ermittelten rPFS-Ergebnissen überein.
Die Wirksamkeitsergebnisse von TALAPRO-2 sind in Tabelle 7 und in Abbildung 4 dargestellt.
Tabelle 7. Zusammenfassung der Wirksamkeitsergebnisse—TALAPRO-2 (mCRPC)* | ||
Talazoparib + Enzalutamid | Placebo + Enzalutamid | |
rPFS gemäß BICR | n = 402 | n = 403 |
Ereignisse, Anzahl (%) | 151 (37,6) | 191 (47,4) |
Median (95 %-KI), Monate | NR (27,5; NR) | 21,9 (16,6; 25,1) |
Hazard Ratio (95 %-KI)a | 0,627 (0,506; 0,777) | |
Zweite Interimsanalyse zum OS | ||
Ereignisse, Anzahl (%) | 156 (38,8) | 174 (43,2) |
Median (95 %-KI), Monate | NR (37,3; NR) | 38,2 (34,1; 43,1) |
Hazard Ratio (95 %-KI)a | 0,837 (0,674; 1,040) | |
Abkürzungen: BICR = verblindete unabhängige zentrale Beurteilung (blinded independent central review); KI = Konfidenzintervall; CSPC = kastrationssensitives Prostatakarzinom (castration resistant prostate cancer); HRR = homologe Rekombinationsreparatur; mCRPC = metastasiertes kastrationsresistentes Prostatakarzinom (metastatic castration-resistent prostate cancer); n = Anzahl Patienten; NHT = neuartige Hormontherapie; NR = nicht erreicht (not reached); OS = Gesamtüberleben (overall survival); rPFS = radiologisches progressionsfreies Überleben (radiographic progression-free survival). | ||
Tabelle 8. Zusammenfassung der Wirksamkeitsergebnisse der Subgruppenanalyse– TALAPRO-2 (mCRPC)* | |||
Talazoparib + Enzalutamid | Placebo + Enzalutamid | ||
Subgruppen-HRRm-Analysena | |||
HRRm | n = 85 | n = 82 | |
rPFS gemäß BICR | |||
Ereignisse, Anzahl (%) | 37 (43,5) | 49 (59,7) | |
Median (95 %-KI), Monate | 27,9 (16,8; NR) | 13,8 (10,9; 19,5) | |
Hazard Ratio (95 %-KI)b | 0,424 (0,275; 0,653) | ||
Zweite Interimsanalyse zum OS | |||
Ereignisse, Anzahl (%) | 30 (35,3) | 41 (50,0) | |
Median (95 %-KI), Monate | 41,9 (36,4; NR) | 30,8 (25,6; 38,8) | |
Hazard Ratio (95 %-KI)b | 0,516 (0,320; 0,831) | ||
Keine HRR-Mutationen | n = 207 | n = 219 | |
rPFS gemäß BICR | |||
Ereignisse, Anzahl (%) | 73 (35,3) | 95 (43,4) | |
Median (95 %-KI), Monate | NR (25,8; NR) | 22,4 (16,6, NR) | |
Hazard Ratio (95 %-KI)b | 0,695 (0,511; 0,944) | ||
Zweite Interimsanalyse zum OS | |||
Ereignisse, Anzahl (%) | 82 (39,6) | 96 (43,8) | |
Median (95 %-KI), Monate | NR (33; NR) | 38 (33,9; NR) | |
Hazard Ratio (95 %-KI)b | 0,880 (0,654; 1,182) | ||
Subgruppen-BRCAm-Analysena | |||
BRCAm | n = 27 | n = 32 | |
rPFS gemäß BICR | |||
Ereignisse, Anzahl (%) | 8 (29,6) | 22 (68,7) | |
Median (95 %-KI), Monate | NR (16,8; NR) | 11 (7,4; 24,6) | |
Hazard Ratio (95 %-KI)b | 0,232 (0,101; 0,529) | ||
Zweite Interimsanalyse zum OS | |||
Ereignisse, Anzahl (%) | 12 (44,4) | 18 (56,3) | |
Median (95 %-KI), Monate | 41,9 (24,9; NR) | 26,1 (15,2; NR) | |
Hazard Ratio (95 %-KI)b | 0,558 (0,263; 1,187) | ||
Abkürzungen: BICR = verblindete unabhängige zentrale Beurteilung (blinded independent central review); BRCAm = mutiertes Brustkrebs-Gen (breast cancer gene mutated); KI = Konfidenzintervall; CSPC = kastrationssensitives Prostatakarzinom (castration resistant prostate cancer); ctDNA = zirkulierende Tumor-DNA (circulating tumour DNA); HRRm = mutiertes homologes Rekombinationsreparatur(HHR)-Gen (homologous recombination repair gene mutated); mCRPC = metastasiertes kastrationsresistentes Prostatakarzinom (metastatic castration-resistent prostate cancer); n = Anzahl Patienten; NHT = neuartige Hormontherapie; NR = nicht erreicht (not reached); OS = Gesamtüberleben (overall survival); rPFS = radiologisches progressionsfreies Überleben (radiographic progression-free survival). | |||
Abbildung 4. Kaplan-Meier-Plot des rPFS gemäß BICR – TALAPRO-2 (mCRPC)
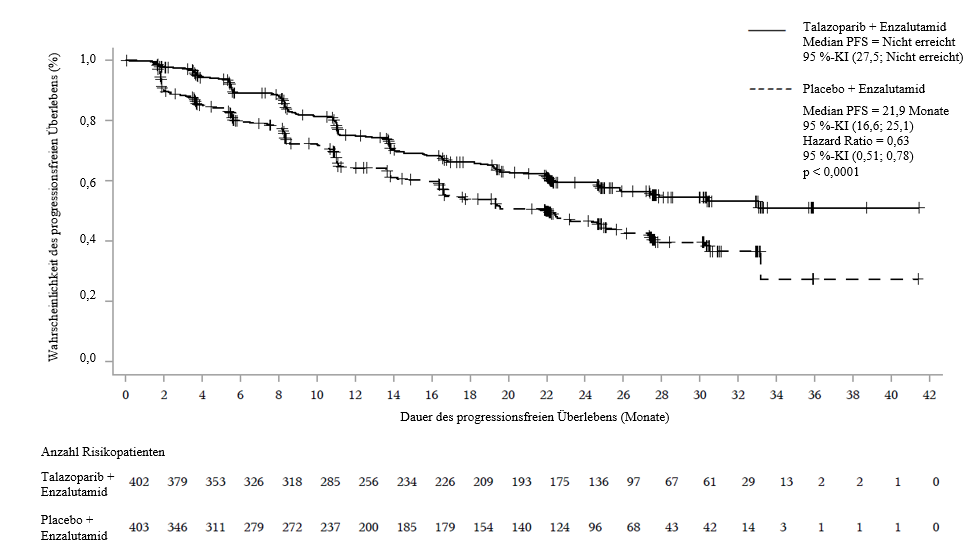
Abkürzungen: BICR = verblindete unabhängige zentrale Beurteilung (blinded independent central review); KI = Konfidenzintervall; mCRPC = metastasiertes kastrationsresistentes Prostatakarzinom (metastatic castration-resistent prostate cancer); PFS = progressionsfreies Überleben (progression-free survival); rPFS = radiologisches progressionsfreies Überleben (radiographic progression-free survival).
Abbildung 5. Forest-Plot der rPFS-Analysen für wichtige Subgruppen – TALAPRO-2 (mCRPC)
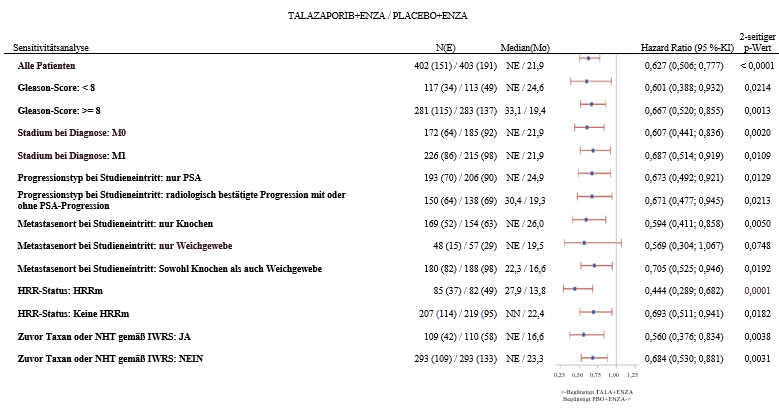
Abkürzungen: KI = Konfidenzintervall; ctDNA = zirkulierende Tumor-DNA; ENZA = Enzalutamid; HRR = homologe Rekombinationsreparatur; HRRm = homologe Rekombinationsreparatur-Genmutation; IWRS = interaktives Web-basiertes Dialogsystem (interactive web response system); mCRPC = metastasiertes kastrationsresistentes Prostatakarzinom (metastatic castration-resistent prostate cancer); n = Anzahl Teilnehmer; NE = nicht auswertbar/nicht erreicht (not evaluable/not reached); NHT = neuartige Hormontherapie; PBO = Placebo; PSA = prostataspezifisches Antigen; rPFS = radiologisches progressionsfreies Überleben (radiographic progression-free survival); SE = Studieneintritt; TALA = Talazoparib; w/o = ohne (without).
Hazard-Ratio für alle Patienten basierte auf einem Cox Modell, stratifiziert nach Radomisierungsstratifizierungsfaktoren. Für alle Subgruppen basierte das Hazard-Ratio auf einem nicht stratifiziertem Cox Model mit der Behandlung als einziger Kovariate. Ein Hazard-Ratio < 1 begünstigt Talazoparib.
HRR Status ist abgeleitet basierend auf Ergebnissen aus einer prospektiven Tumorgewebeanalyse und auf Ergebnissen aus einer prospektiven Untersuchung auf ctDNA im Blut.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für Talazoparib eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in der Behandlung von Brustkrebs und Prostatakrebs gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Die Talazoparib-Exposition stieg im Allgemeinen bei einer Dosierung im Bereich von 0,025 mg bis 2 mg nach täglicher Verabreichung mehrerer Dosen dosisproportional an. Nach wiederholter täglicher Verabreichung von 1 mg Talazoparib als Monotherapie an Brustkrebs-Patienten lagen die geometrisch gemittelte (% Variationskoeffizient [VK%]) Fläche unter der Plasmakonzentrations-Zeit-Kurve (AUC) und maximale Plasmakonzentration (Cmax) von Talazoparib im Steady State im Bereich 126 (107) ng•hr/ml bis 208 (37) ng•hr/ml bzw. 11 (90) ng/ml bis 19 (27) ng/ml. Nach peroraler Verabreichung von 0,5 mg Talazoparib einmal täglich in Kombination mit Enzalutamid bei mCRPC-Patienten lag der geometrische Mittelwert (VK%) der Ctrough im Steady State über alle Besuchstermine hinweg zwischen 3,29 und 3,68 ng/ml (45 bis 48 %). Dies ist ähnlich dem beobachteten Wert von 3,53 ng/ml (61 %) unter einer Talazoparib-Monotherapie mit 1 mg einmal täglich bei Brustkrebspatienten. Nach wiederholter täglicher Dosierung erreichten die Talazoparib-Plasmakonzentrationen den Steady State bei alleiniger Verabreichung innerhalb von 2 bis 3 Wochen und etwa innerhalb von 9 Wochen bei gleichzeitiger Verabreichung mit Enzalutamid. Bei wiederholter einmal täglicher Einnahme von 1 mg als Monotherapie akkumuliert Talazoparib mit einem medianen Akkumulationsverhältnis von 2,3 bis 5,2. Talazoparib ist ein Substrat der P‑gp- und BCRP‑Transporter.
Resorption
Nach Einnahme von Talazoparib betrug die mediane Zeit bis zum Erreichen der Cmax (Tmax) im Allgemeinen 1 bis 2 Stunden. Eine Studie zur absoluten Bioverfügbarkeit beim Menschen wurde nicht durchgeführt. Basierend auf Daten zur Ausscheidung über den Urin liegt die absolute Bioverfügbarkeit jedoch bei mindestens 41 % mit einer resorbierten Fraktion von mindestens 69 % (siehe Elimination). Es wird keine signifikante Auswirkung säurereduzierender Arzneimittel auf die Talazoparib-Exposition erwartet, da Talazoparib bei allen pH‑Werten zwischen 1 und 6,8 ausreichend löslich ist. 28 % der Patienten in der pivotalen Studie nahmen säurereduzierende Arzneimittel ein, vor allem Protonenpumpenhemmer.
Auswirkung der Nahrung
Eine Nahrungsaufnahme verringerte die Resorptionsrate, nicht jedoch das Ausmaß der Resorption von Talazoparib. Nach Einnahme einer Einzeldosis Talazoparib mit fettreicher, kalorienreicher Nahrung (etwa 827 Kalorien, 57 % Fett) verringerte sich die mittlere Cmax von Talazoparib um etwa 46 %, die mediane Tmax verzögerte sich von 1 auf 4 Stunden, während die AUCinf nicht betroffen war. Vor dem Hintergrund dieser Ergebnisse kann Talzenna mit oder ohne Nahrung eingenommen werden (siehe Abschnitt 4.2).
Verteilung
Das populationsgemittelte apparente Verteilungsvolumen (Vss/F) für Talazoparib betrug 420 l. In vitro wird Talazoparib zu etwa 74 % an Plasmaproteine gebunden, ohne Konzentrationsabhängigkeit über einen Konzentrationsbereich von 0,01 µM bis 1 µM. Eine Nieren- oder Leberinsuffizienz scheint sich nicht auf die Proteinbindung von Talazoparib auszuwirken, da in vivo bei sich verschlechternder Nieren- oder Leberfunktion keine offensichtliche Tendenz der mittleren Talazoparib-Fraktion des ungebundenen Wirkstoffs (fu) im menschlichen Plasma festzustellen war.
Biotransformation
Talazoparib unterliegt einer minimalen hepatischen Metabolisierung beim Menschen. Nach oraler Verabreichung einer Einzeldosis von 1 mg [14C] Talazoparib an Menschen wurden keine wichtigen zirkulierenden Metaboliten im Plasma identifiziert, und Talazoparib war die einzige im Plasma zirkulierende medikamentenabgeleitete Einheit. Keine Metaboliten, die jeweils mehr als 10 % der verabreichten Dosis ausmachten, wurden in Urin oder Fäzes wiedergefunden.
In vitro war Talazoparib kein Inhibitor von Cytochrom (CYP)1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 oder CYP3A4/5 und kein Induktor von CYP1A2, CYP2B6 oder CYP3A4 bei klinisch relevanten Konzentrationen.
In vitro hemmte Talazoparib keinen der wichtigen intestinalen, hepatischen oder renalen Membrantransporter (P-gp, BCRP, organisches Anionen-transportierendes Polypeptid [OATP]1B1, OATP1B3, organischer Kationentransporter [OCT]1, OCT2, organischer Anionentransporter [OAT]1, OAT3, Gallensalzexportpumpe [BSEP], Multidrug And Toxin Extrusion [MATE]1 und MATE2-K) bei klinisch relevanten Konzentrationen.
In vitro hemmte Talazoparib keine der wichtigsten Uridindiphosphat-Glucuronosyltransferase(UGT)-Isoformen (1A1, 1A4, 1A6, 1A9, 2B7, und 2B15) bei klinisch relevanten Konzentrationen.
Elimination
Talazoparib wird unverändert hauptsächlich über die Nieren ausgeschieden (passive Filtration und aktive Sekretion). P-gp ist wahrscheinlich an der aktiven renalen Sekretion von Talazoparib beteiligt. Die mittlere (± Standardabweichung) terminale Plasma-Halbwertszeit von Talazoparib betrug 90 (± 58) Stunden und die populationsgemittelte (intersubjektive Variabilität) apparente orale Clearance (CL/F) 6,5 (31 %) l/h bei Krebspatienten. Bei 6 Patientinnen, die eine Einzeldosis [14C] Talazoparib einnahmen, wurden im Mittel 69 % (± 8,6 %) bzw. 20 % (± 5,5 %) der insgesamt verabreichten radioaktiven Dosis in Urin bzw. Fäzes wiedergefunden. Unverändertes Talazoparib wurde hauptsächlich (55 % der verabreichten Dosis) über den Urin ausgeschieden. Über die Fäzes wurden 14 % des unveränderten Talazoparibs ausgeschieden.
Besondere Patientengruppen
Alter, Geschlecht und Körpergewicht
In einer populationspharmakokinetischen Analyse der Daten von 490 Krebspatienten, die Talazoparib als Monotherapie mit 1 mg täglich erhielten, wurden die Auswirkungen von Alter (zwischen 18 und 88 Jahren), Geschlecht (53 Männer und 437 Frauen) und Körpergewicht (zwischen 35,7 kg und 162 kg) auf die Pharmakokinetik (PK) von Talazoparib untersucht. Die Ergebnisse zeigten, dass Alter, Geschlecht und Körpergewicht keine klinisch relevanten Auswirkungen auf die PK von Talazoparib hatten.
Ethnische Zugehörigkeit
Basierend auf einer populationspharmakokinetischen Analyse der Daten von 490 Patienten, die Talazoparib als Monotherapie mit 1 mg täglich erhielten, davon 41 Asiaten und 449 nicht-Asiaten (361 Kaukasier, 16 Schwarze, 9 Andere und 63 ohne Angabe), war die CL/F von Talazoparib bei asiatischen Patienten höher als bei nicht‑asiatischen Patienten. Dies führte zu einer um 19 % niedrigeren Exposition (AUC) bei asiatischen Patienten.
Kinder und Jugendliche
Die pharmakokinetischen Eigenschaften von Talazoparib wurden bei Patienten unter 18 Jahren nicht bewertet.
Niereninsuffizienz
Talazoparib-Monotherapie
Daten aus einer PK-Studie mit Patienten mit Krebserkrankungen im fortgeschrittenen Stadium und Niereninsuffizienz verschiedener Grade wiesen darauf hin, dass sich die Gesamtexposition gegenüber Talazoparib (AUC0-24) nach mehreren Dosen Talazoparib einmal täglich bei Patienten mit mittelschwerer (eGFR 30 – < 60 ml/min) bzw. schwerer (eGFR < 30 ml/min) Niereninsuffizienz im Vergleich zu Patienten mit normaler Nierenfunktion (eGFR ≥ 90 ml/min) um jeweils 92 % bzw. 169 % erhöhte. Die Cmax von Talazoparib erhöhte sich bei Patienten mit mittelschwerer und schwerer Niereninsuffizienz im Vergleich zu Patienten mit normaler Nierenfunktion um jeweils 90 % bzw. 107 %. Bei Patienten mit leichter Niereninsuffizienz (eGFR 60 – < 90 ml/min) und Patienten mit normaler Nierenfunktion war die Talazoparib-Exposition ähnlich. Basierend auf einer populationspharmakokinetischen Analyse an 490 Patienten, von denen 132 Patienten eine leichte Niereninsuffizienz (60 ml/min ≤ CrCl < 90 ml/min), 33 Patienten eine mittelschwere Niereninsuffizienz (30 ml/min ≤ CrCl < 60 ml/min) und 1 Patient eine schwere Niereninsuffizienz (CrCl < 30 ml/min) aufwiesen, verringerte sich zudem die CL/F von Talazoparib um jeweils 14 % bzw. 37 %, entsprechend einer Erhöhung der AUC um 17 % bzw. 59 %, bei Patienten mit leichter bzw. mittelschwerer Niereninsuffizienz im Vergleich zu Patienten mit normaler Nierenfunktion (CrCl ≥ 90 ml/min). Bei Hämodialyse‑Patienten wurden die pharmakokinetischen Eigenschaften von Talazoparib nicht untersucht (siehe Abschnitt 4.2).
Gleichzeitige Verabreichung von Talazoparib mit Enzalutamid
Basierend auf einer populationspharmakokinetischen Analyse an 412 mCRPC-Patienten, die Talazoparib in Kombination mit Enzalutamid erhielten, hatten 152 Patienten eine leichte Niereninsuffizienz (60 ml/min ≤ CrCl < 90 ml/min), 72 Patienten eine mittelschwere Niereninsuffizienz (30 ml/min ≤ CrCl < 60 ml/min) und 2 Patienten eine schwere Niereninsuffizienz (CrCl < 30 ml/min). Die CL/F von Talazoparib war bei Patienten mit leichter bzw. mittelschwerer Niereninsuffizienz um 8 % bzw. 27 % reduziert, was einem Anstieg der AUC um 9 % bzw. 37 % im Vergleich zu Patienten mit normaler Nierenfunktion entspricht. Die PK von Talazoparib wurde bei Hämodialyse-Patienten nicht untersucht (siehe Abschnitt 4.2).
Leberinsuffizienz
Talazoparib-Monotherapie
Basierend auf einer populationspharmakokinetischen Analyse an 490 Patienten, die Talazoparib als Monotherapie mit 1 mg täglich erhielten und von denen 118 Patienten eine leichte Leberinsuffizienz aufwiesen (Gesamtbilirubin ≤ 1,0 × ULN und AST > ULN oder Gesamtbilirubin > 1,0 bis 1,5 × ULN und beliebige AST), zeigte eine leichte Leberinsuffizienz keinen Effekt auf die PK von Talazoparib. In einer PK-Studie wurde die PK von Talazoparib bei Patienten mit normaler Leberfunktion, leichter Leberinsuffizienz, mittelschwerer Leberinsuffizienz (Gesamtbilirubin > 1,5 bis 3,0 × ULN und beliebige AST) oder schwerer Leberinsuffizienz (Gesamtbilirubin > 3,0 × ULN und beliebige AST) untersucht. Die auf Daten aus dieser PK-Studie basierende populationspharmakokinetische Analyse zeigte, dass eine leichte, mittelschwere oder schwere Leberinsuffizienz keine signifikanten Auswirkungen auf die PK von Talazoparib hatte (siehe Abschnitt 4.2).
Gleichzeitige Verabreichung von Talazoparib mit Enzalutamid
Die PK von Talazoparib in Kombination mit Enzalutamid wurde bei Patienten mit Leberinsuffizienz nicht untersucht (siehe Abschnitt 4.2).
Karzinogenität
Karzinogenitätsstudien mit Talazoparib wurden nicht durchgeführt.
Genotoxizität
Talazoparib erwies sich in einem bakteriellen Rückmutationstest (Ames) als nicht mutagen. Talazoparib war im In‑vitro‑Chromosomenaberrationstest an humanen Lymphozyten im peripheren Blut und In‑vivo‑Mikrokerntest im Knochenmark von Ratten bei einer Exposition im humantherapeutischen Dosisbereich klastogen. Diese Klastogenität entspricht der pharmakologisch bedingten genomischen Instabilität von Talazoparib und weist auf eine potenzielle Genotoxizität beim Menschen hin.
Toxizität bei wiederholter Gabe
In Studien zur chronischen Toxizität an Ratten und Hunden waren die wichtigsten Befunde bei subtherapeutischer Exposition eine Hypozellularität des Knochenmarks mit dosisabhängiger Abnahme der hämatopoetischen Zellen, eine Depletion des lymphatischen Gewebes in mehreren Organen und eine Atrophie und/ oder degenerative Veränderungen von Hoden, Nebenhoden und Samenkanälchen. Weitere Ergebnisse bei höheren Expositionen umfassten einen dosisabhängigen Anstieg von Apoptosen/Nekrosen im Magen-Darm-Trakt, in der Leber und in den Ovarien. Die meisten der histopathologischen Befunde waren im Allgemeinen reversibel. Der Befund an den Hoden war jedoch 4 Wochen nach dem Absetzen der Behandlung nur teilweise reversibel. Diese Ergebnisse stehen im Einklang mit der pharmakologischen Wirkung von Talazoparib und dem Verteilungsmuster im Gewebe.
Entwicklungstoxizität
In einer Studie zur embryofetalen Entwicklung an Ratten verursachte Talazoparib embryofetale Sterblichkeit, fetale Missbildungen (verringertes Vorwölben der Augen, kleines Auge, gespaltenes Brustbein, fusionierter Halswirbelbogen) und Skelettveränderungen bei einer mütterlichen Exposition (systemische AUC0‑24) von nur etwa dem 0,09‑fachen der humantherapeutischen Exposition bei der empfohlenen Dosis.
Kapselinhalt
Mikrokristalline Cellulose, Siliciumdioxid-beschichtet
Kapselhülle 0,1 mg
Hypromellose
Titandioxid (E 171)
Kapselhülle 0,25 mg
Hypromellose
Eisen(III)‑hydroxid‑oxid × H2O (E 172)
Titandioxid (E 171)
Kapselhülle 0,35 mg
Hypromellose
Eisen(III)‑hydroxid‑oxid × H2O (E 172)
Titandioxid (E 171)
Kapselhülle 0,5 mg
Hypromellose
Eisen(III)‑oxid (E 172)
Titandioxid (E 171)
Kapselhülle 1 mg
Hypromellose
Eisen(III)‑oxid (E 172)
Eisen(III)‑hydroxid‑oxid × H2O (E 172)
Titandioxid (E 171)
Drucktinte
Schellack (E 904)
Propylenglycol (E 1520)
Ammoniakwasser (E 527)
Eisen(II,III)‑oxid (E 172)
Kaliumhydroxid (E 525)
Nicht zutreffend.
4 Jahre
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Talzenna 0,1 mg Hartkapseln
Polyethylen hoher Dichte (HDPE)‑Flasche mit Polypropylen(PP)‑Verschluss mit Wärmeinduktionsversiegelung. Packungsgröße: Umkartons mit 30 Kapseln in einer HDPE‑Flasche.
Talzenna 0,25 mg Hartkapseln
Polyethylen hoher Dichte (HDPE)‑Flasche mit Polypropylen (PP)‑Verschluss mit Wärmeinduktionsversiegelung. Packungsgröße: Umkartons mit 30 Kapseln in einer HDPE‑Flasche.
Perforierte Blisterpackung zur Abgabe von Einzeldosen aus Polyvinylchlorid/ Polyvinylidenchlorid (PVC/ PVdC) mit Aluminiumabziehfolie. Packungsgrößen: Umkartons mit 30 × 1 Kapsel oder 60 × 1 Kapsel oder 90 × 1 Kapsel in Blisterpackungen zur Abgabe von Einzeldosen.
Talzenna 0,35 mg Hartkapseln
Polyethylen hoher Dichte (HDPE)-Flasche mit Polypropylen (PP)Verschluss mit Wärmeinduktionsversiegelung. Packungsgröße: Umkartons mit 30 Kapseln in einer HDPE-Flasche.
Talzenna 0,5 mg Hartkapseln
Polyethylen hoher Dichte (HDPE)‑Flasche mit Polypropylen (PP)‑Verschluss mit Wärmeinduktionsversiegelung. Packungsgröße: Umkartons mit 30 Kapseln in einer HDPE‑Flasche.
Talzenna 1 mg Hartkapseln
Polyethylen hoher Dichte (HDPE)‑Flasche mit Polypropylen (PP)‑Verschluss mit Wärmeinduktionsversiegelung. Packungsgröße: Umkartons mit 30 Kapseln in einer HDPE‑Flasche.
Perforierte Blisterpackung zur Abgabe von Einzeldosen aus Polyvinylchlorid/ Polyvinylidenchlorid (PVC/ PVdC) mit Aluminiumabziehfolie. Packungsgröße: Umkartons mit 30 × 1 Kapsel in Blisterpackungen zur Abgabe von Einzeldosen.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Brüssel
Belgien
Talzenna 0,1 mg Hartkapseln
EU/1/19/1377/007
Talzenna 0,25 mg Hartkapseln
EU/1/19/1377/001
EU/1/19/1377/002
EU/1/19/1377/003
EU/1/19/1377/004
Talzenna 0,35 mg Hartkapseln
EU/1/19/1377/008
Talzenna 0,5 mg Hartkapseln
EU/1/19/1377/009
Talzenna 1 mg Hartkapseln
EU/1/19/1377/005
EU/1/19/1377/006
Datum der Erteilung der Zulassung: 20. Juni 2019
Datum der letzten Verlängerung der Zulassung: 15. April 2024
Juni 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.

VERKAUFSABGRENZUNG IN DEUTSCHLAND
Verschreibungspflichtig
REZEPTPFLICHT/APOTHEKENPFLICHT IN ÖSTERREICH
Rezept- und apothekenpflichtig, wiederholte Abgabe verboten
PACKUNGSGRÖSSEN IN DEUTSCHLAND
TALZENNA 0,1 mg, TALZENNA, 0,25 mg, TALZENNA 0,35 mg, TALZENNA 0,5 mg, TALZENNA 1 mg: Flaschen mit 30 Hartkapseln (N1)
PACKUNGSGRÖSSEN IN ÖSTERREICH
TALZENNA 0,1 mg, TALZENNA, 0,25 mg, TALZENNA 1 mg: Flaschen mit 30 Hartkapseln
REPRÄSENTANT IN DEUTSCHLAND
PFIZER PHARMA GmbH
Friedrichstr. 110
10117 Berlin
Tel.: 030 550055-51000
Fax: 030 550054-10000
REPRÄSENTANT IN ÖSTERREICH
Pfizer Corporation Austria Ges.m.b.H.
Floridsdorfer Hauptstraße 1
A-1210 Wien
Tel.: +43 (0)1 521 15-0