
Filsuvez Gel
1 g Gel enthält 100 mg raffinierten Trockenextrakt aus Rinde von Betula pendula Roth, Betula pubescens Ehrh. sowie Hybriden beider Arten (äquivalent zu 0,5 - 1,0 g Birkenrinde), quantifiziert auf 84 - 95 mg Triterpene als Summe berechnet aus Betulin, Betulinsäure, Erythrodiol, Lupeol und Oleanolsäure; Auszugsmittel: n‑Heptan.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Gel
Farbloses bis leicht gelbliches, opalisierendes, nicht-wässriges Gel.
Filsuvez wird angewendet bei oberflächlichen Wunden im Zusammenhang mit dystropher und junktionaler Epidermolysis bullosa (EB) bei Patienten ab 6 Monaten.
Dosierung
Das Gel sollte entweder mit einer Dicke von etwa 1 mm direkt auf die Wundoberfläche aufgetragen und mit einer sterilen, nicht haftenden Wundauflage abgedeckt werden oder so auf die Wundauflage aufgetragen werden, dass das Gel direkten Kontakt zur Wunde hat. Das Gel sollte nicht sparsam aufgetragen werden. Es sollte nicht eingerieben werden. Das Gel sollte bei jedem Verbandwechsel erneut aufgetragen werden. Die in klinischen Studien behandelte Gesamtwundfläche betrug maximal 5 300 cm2 bei einer medianen Gesamtwundfläche von 735 cm2. Wenn die Symptome nach der Anwendung fortbestehen oder sich verschlimmern, oder wenn Wundkomplikationen auftreten, sollte der Zustand des Patienten vor der Fortsetzung der Behandlung umfassend klinisch beurteilt und danach regelmäßig neu bewertet werden.
Besondere Patientengruppen
Patienten mit eingeschränkter Nieren- oder Leberfunktion
Es wurden keine Studien mit Filsuvez bei Patienten mit eingeschränkter Nieren- oder Leberfunktion durchgeführt. Für Patienten mit eingeschränkter Nieren- oder Leberfunktion sind weder Dosisanpassungen noch besondere Maßnahmen vorgesehen (siehe Abschnitt 5.2).
Ältere Patienten
Eine Dosisanpassung ist nicht erforderlich.
Kinder und Jugendliche
Die Dosierung bei Kindern und Jugendlichen (6 Monate oder älter) ist die Gleiche wie bei Erwachsenen. Die Sicherheit und Wirksamkeit von Filsuvez bei Kindern im Alter unter 6 Monaten ist bisher noch nicht erwiesen.
Es liegen keine Daten vor.
Art der Anwendung
Nur zur Anwendung auf der Haut.
Filsuvez sollte auf die gereinigten Wunden aufgetragen werden. Dieses Arzneimittel ist nicht zur Anwendung am Auge bestimmt und sollte nicht auf Schleimhäute aufgetragen werden.
Jede Tube ist nur zum einmaligen Gebrauch bestimmt. Die Tube sollte nach dem Gebrauch entsorgt werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Überempfindlichkeit
Bei Patienten, die mit Filsuvez behandelt wurden, sind Überempfindlichkeitsreaktionen aufgetreten (siehe Abschnitt 4.8). Wenn Anzeichen und Symptome einer lokalen oder systemischen Überempfindlichkeit auftreten, muss Filsuvez sofort abgesetzt und eine geeignete Behandlung eingeleitet werden.
Wundinfektion
Das Gel ist steril. Eine Wundinfektion ist jedoch eine erhebliche und schwerwiegende Komplikation, die während der Wundheilung auftreten kann. Es wird empfohlen, im Fall einer Infektion die Behandlung zu unterbrechen. Eine zusätzliche Standardbehandlung kann erforderlich sein (siehe Abschnitt 4.5). Die Behandlung kann wieder aufgenommen werden, sobald die Infektion abgeklungen ist.
Plattenepithelkarzinom und andere bösartige Hauterkrankungen
Bei Patienten mit dystropher Epidermolysis bullosa (DEB) und junktionaler Epidermolysis bullosa (JEB) besteht möglicherweise ein erhöhtes Risiko für die Entwicklung eines Plattenepithelkarzinoms. Obwohl bisher kein erhöhtes Hautkrebsrisiko im Zusammenhang mit Filsuvez festgestellt wurde, kann ein theoretisch erhöhtes Hautkrebsrisiko im Zusammenhang mit der Anwendung von Filsuvez nicht ausgeschlossen werden. Im Falle der Diagnose eines Plattenepithelkarzinoms oder anderer Hautkrebsarten sollte die Behandlung des betroffenen Bereichs abgebrochen werden.
Anwendung bei dominanter dystropher EB (DDEB) und junktionaler EB (JEB)
Die Menge der klinischen Daten zur Anwendung von Filsuvez bei Patienten mit DDEB und JEB ist begrenzt (siehe Abschnitt 5.1). Der Zustand des Patienten sollte regelmäßig überprüft werden, um den Nutzen einer fortgesetzten Behandlung zu beurteilen.
Birkenpollenallergie
Die Anwendung von Filsuvez bei Personen mit Birkenpollenallergie ist sicher, da die entsprechenden Allergene nicht in diesem Arzneimittel enthalten sind.
Unbeabsichtigter Kontakt mit den Augen
Bei Kontakt mit den Augen sollte das Produkt durch Augenspülung entfernt werden.
Es wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt. Da die systemische Exposition des Hauptbestandteils Betulin nach Anwendung auf der Haut vernachlässigbar ist, ist keine Wechselwirkung mit systemischen Behandlungen zu erwarten. Wechselwirkungen mit topischen Produkten wurden in klinischen Studien nicht untersucht. Andere topische Produkte sollten nicht gleichzeitig mit Filsuvez, sondern, je nach klinischer Notwendigkeit, nacheinander oder abwechselnd angewendet werden.
Schwangerschaft
Bisher liegen keine oder nur sehr begrenzte Erfahrungen mit der Anwendung von Filsuvez bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3). Da die systemische Exposition durch Filsuvez zu vernachlässigen ist, wird davon ausgegangen, dass während einer Schwangerschaft keine Wirkungen auftreten. Filsuvez kann während der Schwangerschaft angewendet werden.
Stillzeit
Es ist nicht bekannt ob Birkenrindenextrakt/Metabolite in die Muttermilch übergehen. Es wird angenommen, dass Filsuvez keine Auswirkungen auf das gestillte Neugeborene/Kind hat, weil die systemische Exposition der stillenden Frau gegenüber Filsuvez vernachlässigbar ist. Filsuvez kann während der Stillzeit angewendet werden, es sei denn, die Behandlung erfolgt im Brustbereich.
Fertilität
Bei männlichen und weiblichen Ratten, denen Birkenrindenextrakt verabreicht wurde, wurden keine nachteiligen Auswirkungen auf die Fertilität beobachtet. Da die systemische Exposition vernachlässigbar ist, sind keine Auswirkungen auf die menschliche Fertilität zu erwarten.
Filsuvez hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten beobachteten Nebenwirkungen waren Wundkomplikationen (bei 11,6 % der Patienten mit EB und 2,9 % der Patienten mit anderen oberflächlichen Wunden (partial thickness wounds, PTW)), Reaktionen an der Applikationsstelle (bei 5,8 % der Patienten mit EB), Wundinfektionen (bei 4,0 % der Patienten mit EB), Pruritus (bei 3,1 % der Patienten mit EB und 1,3 % der Patienten mit anderen PTW), Hautschmerzen (bei 2,5 % der Patienten mit anderen PTW) und Überempfindlichkeitsreaktionen (bei 1,3 % der Patienten mit EB). Es gab keine klinisch relevanten Unterschiede bei den Reaktionen, die bei Patienten mit EB im Vergleich zu Patienten mit anderen PTW berichtet wurden.
Tabellarische Auflistung der Nebenwirkungen
In der folgenden Tabelle sind die Nebenwirkungen nach MedDRA-Systemorganklasse und bevorzugter Bezeichnung aufgeführt. Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Die Häufigkeit der Nebenwirkungen ist wie folgt definiert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
In Tabelle 1 sind alle in klinischen Studien berichteten Nebenwirkungen aufgeführt.
Tabelle 1: Nebenwirkungen
Systemorganklasse | Sehr häufig | Häufig | Gelegentlich |
Infektionen und parasitäre Erkrankungen | Wundinfektionen | ||
Erkrankungen des Immunsystems | Überempfindlichkeitsreaktionen* | ||
Erkrankungen der Haut und des Unterhautgewebes | Wundkomplikation* | Pruritus | |
Dermatitisa | |||
Ausschlag mit Juckreiza | |||
Purpuraa | |||
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Reaktionen an der Applikationsstelle* | Schmerza | |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen | Wundkomplikation*a | Wundsekretion |
* Siehe Beschreibung ausgewählter Nebenwirkungen
a Nebenwirkungen, die in Studien bei Patienten mit Verbrennungswunden vom Grad 2a oder Spalthauttransplantaten beobachtet wurden
Beschreibung ausgewählter Nebenwirkungen
Überempfindlichkeit
In klinischen Studien bei Patienten mit EB wurden häufig überempfindlichkeitsartige Reaktionen beobachtet. Zu diesen Reaktionen gehören Ausschlag, Urtikaria und Ekzem, die bei 1,3 % der Patienten leicht und bei 0,4 % der Patienten schwer waren. Spezifische Empfehlungen siehe Abschnitt 4.4.
Reaktionen an der Applikationsstelle
Leichte oder mäßige Reaktionen an der Applikationsstelle sind häufig und umfassen Schmerzen an der Applikationsstelle und Pruritis an der Applikationsstelle.
Wundkomplikation
In Studien an Patienten mit Epidermolysis bullosa umfassten Wundkomplikationen verschiedene Arten lokaler Komplikationen wie eine Vergrößerung der Wundfläche, eine Wiedereröffnung der Wunde, Wundschmerz und Wundblutung.
In Studien an Patienten mit Verbrennungswunden oder Spalthauttransplantaten umfassten Wundkomplikationen verschiedene Arten lokaler Komplikationen wie Komplikationen nach dem Eingriff, Wundnekrose, Wundsekretion, verzögerte Heilung oder Wundentzündung.
Kinder und Jugendliche
70 % (n = 156) der randomisierten Patienten in der Zulassungsstudie (siehe Abschnitt 5.1) waren unter 18 Jahre alt, das mediane Alter lag bei 12 Jahren. 8 % (n = 17) der Patienten waren unter 4 Jahre alt und 2 Patienten waren unter 1 Jahr alt. Die in der Gesamtpopulation beobachteten Nebenwirkungen waren vergleichbar mit denen, die in der pädiatrischen Population beobachtet wurden.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: http://www.bfarm.de
anzuzeigen.
Eine Überdosierung von Filsuvez ist unwahrscheinlich. Bei einer täglichen Anwendung von maximal 69 g über mehr als 90 Tage wurde kein Fall einer Überdosierung gemeldet.
Zur versehentlichen Einnahme von Filsuvez liegen keine Daten vor. Die weitere Behandlung sollte nach klinischer Indikation erfolgen.
Pharmakotherapeutische Gruppe: Zubereitung zur Behandlung von Wunden und Geschwüren, Andere Wundbehandlungsmittel; ATC-Code: D03AX13.
Wirkmechanismus und pharmakodynamische Wirkungen
Untersuchungen an Zellkulturen mit primären humanen Keratinozyten und Fibroblasten sowie Ex vivo-Studien an Schweinehaut zeigen, dass der Extrakt einschließlich des Hauptbestandteils Betulin Entzündungsmediatoren moduliert und mit der Aktivierung intrazellulärer Signalwege verbunden ist, von denen bekannt ist, dass sie an der Differenzierung und Migration von Keratinozyten sowie an Wundheilung und -verschluss beteiligt sind.
Der genaue Wirkmechanismus von Filsuvez auf die Wundheilung ist nicht bekannt.
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit und Sicherheit von Filsuvez bei der Behandlung von oberflächlichen Wunden im Zusammenhang mit hereditärer EB wurde in einer zulassungsrelevanten globalen, randomisierten, doppelblinden, kontrollierten Phase-3-Studie an Erwachsenen und Kindern untersucht (Studie BEB-13; EASE). Patienten mit dystropher EB (DEB) und junktionaler EB (JEB) wurden im Verhältnis 1:1 randomisiert und erhielten Filsuvez (n = 109) oder ein verblindetes Kontrollgel (bestehend aus raffiniertem Sonnenblumenöl, gelbem Bienenwachs und Carnaubawachs) (n = 114); sie wurden angewiesen, das Prüfpräparat 90 Tage lang bei jedem Wechsel der Wundauflage (alle 1 bis 4 Tage) in einer etwa 1 mm dicken Schicht auf alle Wunden aufzutragen. Bei der Randomisierung wurde eine Wunde vom Prüfer als Zielwunde für die Beurteilung des primären Wirksamkeitsendpunkts ausgewählt. Die Zielwunde war als eine oberflächliche Wunde mit einer Fläche von 10-50 cm2 definiert, die seit mindestens 21 Tagen bis höchstens 9 Monate vor dem Screening bestand. Der primäre Endpunkt war der Anteil der Patienten, bei denen sich die Zielwunde bis zum 45. Tag der 90-tägigen Doppelblindphase (DBP) der Studie erstmals vollständig geschlossen hatte. Nach Abschluss der DBP traten die Patienten in eine 24-monatige Open-Label-Phase (OLP) der Studie ein, in der alle Wunden mit Filsuvez behandelt wurden.
Das mediane Alter der 223 randomisierten Patienten betrug 12 Jahre (Spanne: 6 Monate bis 81 Jahre), 70 % der Patienten waren unter 18 Jahre alt und 8 % der Patienten waren unter 4 Jahre alt. 60 % der randomisierten Patienten waren männlich. Von diesen 223 Patienten hatten 195 eine DEB, davon 175 eine rezessive DEB (RDEB) und 20 eine dominante DEB (DDEB); 26 Patienten hatten eine JEB. In der DBP trug die Mehrheit der Patienten das Studienarzneimittel entweder täglich oder alle zwei Tage auf alle Wunden auf (zwischen 70 % und 78 %). Für Patienten mit schwarzer Hautfarbe und asiatische Patienten liegen nur begrenzte Daten vor.
Die Ergebnisse, einschließlich des primären Endpunkts, sind in Tabelle 2 dargestellt.
Tabelle 2: Ergebnisse zur Wirksamkeit (Studie BEB-13; 90-Tage-Doppelblindphase, Gesamtkollektiv (full analysis set, FAS))
Wirksamkeitsparameter | Filsuvez | Kontrollgel | p-Wert |
Anteil der Patienten mit erstem vollständigen Verschluss der Zielwunde innerhalb von 45 Tagen | 41,3 % | 28,9 % | 0,013 |
Nach EB-Subtyp | |||
RDEB (n = 175) | 44,0 % | 26,2 % | 0,008 |
DDEB (n = 20) | 50,0 % | 50,0 % | 0,844 |
JEB (n = 26) | 18,2 % | 26,7 % | 0,522 |
Anteil der Patienten mit erstem vollständigen Verschluss der Zielwunde innerhalb von 90 Tagen* | 50,5 % | 43,9 % | 0,296 |
* wichtiger sekundärer Endpunkt
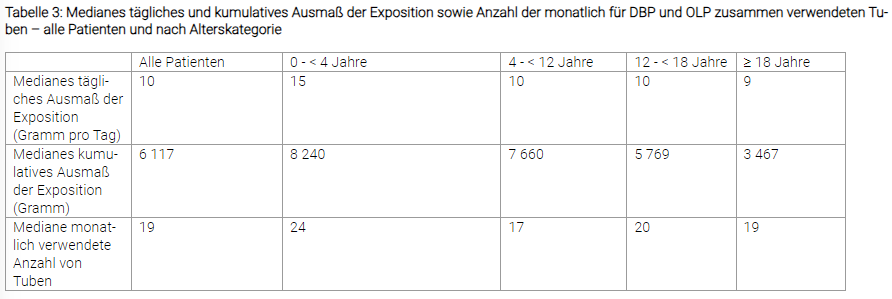
Das mediane tägliche Ausmaß der Exposition für alle Patienten in der DBP und der OLP zusammen ist in Tabelle 3 dargestellt. Die mediane Dauer der Behandlung mit Filsuvez für alle Patienten in der DBP und OLP beträgt 733 Tage mit einem Maximum von 931 Tagen.
Resorption
Die systemische Exposition gegenüber dem Hauptbestandteil Betulin wurde zu Beginn und in regelmäßigen Abständen während der BEB-13-Studie unter Verwendung einer bioanalytischen Methode mit getrockneten Bluttropfen ermittelt. Die Betulinkonzentrationen im venösen Blut lagen bei der großen Mehrheit der Studienteilnehmer unter der Bestimmungsgrenze (10 ng/ml). Bei einer Minderheit der Studienteilnehmer wurden messbare Betulinkonzentrationen im venösen Blut festgestellt, was darauf hindeutet, dass die Resorption von topisch verabreichtem Betulin minimal ist. Diese venösen Blutkonzentrationen, die nicht mehr als 207 ng/ml betrugen, waren mit denen vergleichbar, die bei der Einnahme von Betulin-haltigen Nahrungsmitteln beobachtet wurden.
Verteilung
Die Plasmaproteinbindung von Betulin beträgt > 99,9 %.
Biotransformation
Der in vitro-Metabolismus von Betulin wurde in einer Suspension menschlicher Hepatozyten untersucht, wobei 99 % innerhalb von fünf Stunden vollständig metabolisiert wurden. Der am häufigsten vorkommende Metabolit wurde in vitro durch Oxidation, Methylierung und Sulfatierung gebildet. Drei weitere Metaboliten wurden durch Sulfatierung oder Glucuronidierung gebildet. Es wird davon ausgegangen, dass nicht-CYP-vermittelte Stoffwechselwege die vorherrschende Rolle beim gesamten hepatischen Metabolismus von Betulin spielen (75 %), während die CYP-vermittelten Stoffwechselwege (25 %) hauptsächlich durch das Isoenzym CYP3A4/5 gesteuert werden.
Betulin zeigte eine direkte Hemmung von CYP2C8 (Testsubstrat Amodiaquin) und CYP3A (Testsubstrate Testosteron und Midazolam) mit IC50‑Werten von 0,60 µM (266 ng/ml), 0,17 µM (75 ng/ml) bzw. 0,62 µM (275 ng/ml) in menschlichen Hepatozyten. Darüber hinaus verursachte Betulin eine sehr geringe Induktion von CYP3A4 mRNA (2,7‑fach). Da die systemische Exposition jedoch vernachlässigbar ist, sind keine Wechselwirkungen mit systemischen Behandlungen zu erwarten.
Elimination
Es wurden keine in vivo-Studien zur Elimination durchgeführt.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Anwendung, Reproduktions- und Entwicklungstoxizität und Phototoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Nach einer 4-wöchigen topischen Behandlung mit Filsuvez Gel wurden bei Minischweinen mehrere Reaktionen an der Applikationsstelle beobachtet, darunter entzündliche Wirkungen, eine lympho-histiozytäre Infiltration von Entzündungszellen und eine Epithelhyperplasie. Nach einer 9-monatigen Behandlung der Haut bei Minischweinen wurden bei einigen Tieren epidermale Hyperplasie, orthokeratotische Hyperkeratose, dermale lymphozytäre und/oder neutrophile Infiltration und Pusteln im Stratum corneum beobachtet.
In vitro-Studien zur Genotoxizität waren negativ. Es wurden keine weiteren Studien zu Genotoxizität bzw. Karzinogenität durchgeführt.
Raffiniertes Sonnenblumenöl.
Nicht zutreffend.
4 Jahre.
Nach Anbruch muss das Gel unverzüglich angewendet und nach der Anwendung entsorgt werden.
Nicht über 30 °C lagern.
Weiße, faltbare Aluminiumtube, innen mit Epoxid-Phenolharz beschichtet und mit einer Dichtungsmasse im Falz. Die Tube ist mit einer Aluminiummembran als Originalitätsverschluss versehen und mit einem weißen Polypropylen-Schraubdeckel verschlossen. Die Tube ist in einer Faltschachtel verpackt.
Packungsgrößen:
1, 10 und 30 Tuben mit 23,4 g Gel.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Keine besonderen Anforderungen.
Chiesi Farmaceutici S.p.A.
Via Palermo 26/A
43122 Parma
Italien
Filsuvez Gel, 23,4 g Tube
EU/1/22/1652/002
EU/1/22/1652/004
EU/1/22/1652/005
Datum der Erteilung der Zulassung: 21. Juni 2022
August 2024
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
---------------------------------------------------------------------------------------------------------------------------
Verschreibungspflichtig
Chiesi GmbH
Gasstraße 6
22761 Hamburg
Telefon: 040 89724-0
Telefax: 040 89724-212
E-Mail: info.de@chiesi.com