
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Lojuxta 5 mg Hartkapseln
Lojuxta 10 mg Hartkapseln
Lojuxta 20 mg Hartkapseln
Lojuxta 30 mg Hartkapseln
Lojuxta 40 mg Hartkapseln
Lojuxta 60 mg Hartkapseln
Lojuxta 5 mg Hartkapseln
Jede Hartkapsel enthält Lomitapidmesilat entsprechend 5 mg Lomitapid.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Hartkapsel enthält 70,12 mg Lactose (als Monohydrat) (siehe Abschnitt 4.4).
Lojuxta 10 mg Hartkapseln
Jede Hartkapsel enthält Lomitapidmesilat entsprechend 10 mg Lomitapid.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Hartkapsel enthält 140,23 mg Lactose (als Monohydrat) (siehe Abschnitt 4.4).
Lojuxta 20 mg Hartkapseln
Jede Hartkapsel enthält Lomitapidmesilat entsprechend 20 mg Lomitapid.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Hartkapsel enthält 129,89 mg Lactose (als Monohydrat) (siehe Abschnitt 4.4).
Lojuxta 30 mg Hartkapseln
Jede Hartkapsel enthält Lomitapidmesilat entsprechend 30 mg Lomitapid.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Hartkapsel enthält 194,84 mg Lactose (als Monohydrat) (siehe Abschnitt 4.4).
Lojuxta 40 mg Hartkapseln
Jede Hartkapsel enthält Lomitapidmesilat entsprechend 40 mg Lomitapid.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Hartkapsel enthält 259,79 mg Lactose (als Monohydrat) (siehe Abschnitt 4.4).
Lojuxta 60 mg Hartkapseln
Jede Hartkapsel enthält Lomitapidmesilat entsprechend 60 mg Lomitapid.
Sonstiger Bestandteil mit bekannter Wirkung
Jede Hartkapsel enthält 389,68 mg Lactose (als Monohydrat) (siehe Abschnitt 4.4).
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Hartkapsel.
Lojuxta 5 mg Hartkapseln
Bei der Kapsel handelt es sich um eine Hartkapsel mit einer Länge von 19,4 mm, mit orangefarbenem Oberteil/orangefarbenem Unterteil, mit dem Aufdruck „5 mg“ auf dem Unterteil und „A733“ auf dem Oberteil in schwarzer Tinte.
Lojuxta 10 mg Hartkapseln
Bei der Kapsel handelt es sich um eine Hartkapsel mit einer Länge von 19,4 mm, mit orangefarbenem Oberteil/weißem Unterteil, mit dem Aufdruck „10 mg“ auf dem Unterteil und „A733“ auf dem Oberteil in schwarzer Tinte.
Lojuxta 20 mg Hartkapseln
Bei der Kapsel handelt es sich um eine Hartkapsel mit einer Länge von 19,4 mm, mit weißem Oberteil/weißem Unterteil, mit dem Aufdruck „20 mg“ auf dem Unterteil und „A733“ auf dem Oberteil in schwarzer Tinte.
Lojuxta 30 mg Hartkapseln
Bei der Kapsel handelt es sich um eine Hartkapsel mit einer Länge von 21,6 mm, mit orangefarbenem Oberteil/ gelbem Unterteil, mit dem Aufdruck „30 mg“ auf dem Unterteil und „A733“ auf dem Oberteil in schwarzer Tinte.
Lojuxta 40 mg Hartkapseln
Bei der Kapsel handelt es sich um eine Hartkapsel mit einer Länge von 23,4 mm, mit gelbem Oberteil/weißem Unterteil, mit dem Aufdruck „40 mg“ auf dem Unterteil und „A733“ auf dem Oberteil in schwarzer Tinte.
Lojuxta 60 mg Hartkapseln
Bei der Kapsel handelt es sich um eine Hartkapsel mit einer Länge von 23,4 mm, mit gelbem Oberteil/gelbem Unterteil, mit dem Aufdruck „60 mg“ auf dem Unterteil und „A733“ auf dem Oberteil in schwarzer Tinte.
Lojuxta ist begleitend zu einer fettarmen Diät und anderen lipidsenkenden Arzneimitteln mit oder ohne Low‑Density‑Lipoprotein‑Apherese (LDL‑Apherese) bei erwachsenen Patienten mit homozygoter familiärer Hypercholesterinämie (HoFH) angezeigt.
Die Diagnose HoFH sollte, wenn möglich, genetisch bestätigt werden. Andere Formen primärer Hyperlipoproteinämien sowie sekundäre Ursachen von Hypercholesterinämien (z. B. nephrotisches Syndrom oder Hypothyreose) müssen ausgeschlossen werden.
Die Behandlung mit Lojuxta sollte von einem in der Behandlung von Lipidstörungen erfahrenen Arzt eingeleitet und überwacht werden.
Dosierung
Die empfohlene Anfangsdosis beträgt 5 mg einmal täglich. Nach 2 Wochen kann die Dosis entsprechend dem LDL‑C‑Ansprechen bei einer akzeptablen Sicherheit und Verträglichkeit auf 10 mg und danach in Mindestabständen von 4 Wochen auf 20 mg, 40 mg und die empfohlene Höchstdosis von 60 mg erhöht werden (siehe Abschnitt 4.4).
Die Dosis sollte schrittweise erhöht werden, um die Inzidenz und Schwere gastrointestinaler Nebenwirkungen und erhöhter Aminotransferasen zu minimieren.
Das Auftreten und die Schwere gastrointestinaler Nebenwirkungen im Zusammenhang mit der Anwendung von Lojuxta werden durch die Einhaltung einer fettarmen Diät reduziert. Die Patienten sollten vor Beginn der Behandlung eine Diät beginnen, bei der weniger als 20 % der Energie aus Fett stammen, und diese Diät während der Behandlung fortsetzen. Es sollte eine Diätberatung stattfinden.
Die Patienten sollten den Konsum von Grapefruitsaft und Alkohol vermeiden (siehe Abschnitte 4.4 und 4.5).
Bei Patienten mit einer stabilen Erhaltungsdosis von Lojuxta, die Atorvastatin erhalten, sollte entweder:
ein zeitlicher Abstand von 12 Stunden zwischen den Arzneimitteldosen eingehalten werden
ODER
die Dosis von Lojuxta halbiert werden. Patienten mit einer Dosis von 5 mg sollten bei 5 mg bleiben.
Eine vorsichtige Titrierung kann danach gemäß dem LDL‑C‑Ansprechen und der Sicherheit/Verträglichkeit in Erwägung gezogen werden. Nach dem Absetzen von Atorvastatin sollte die Dosis von Lojuxta gemäß dem LDL‑C‑Ansprechen und der Sicherheit/Verträglichkeit auftitriert werden.
Bei Patienten mit einer stabilen Erhaltungsdosis von Lojuxta, die einen anderen schwachen Cytochroms P450 (CYP3A4)‑Hemmer erhalten, ist ein zeitlicher Abstand von 12 Stunden zwischen den Arzneimitteldosen (von Lojuxta und dem schwachen CYP3A4‑Hemmer) einzuhalten. Zusätzliche Vorsicht ist geboten, wenn mehr als ein schwacher CYP3A4‑Hemmer zusammen mit Lojuxta gegeben wird. Es sollte in Erwägung gezogen werden, die Höchstdosis von Lojuxta gemäß dem gewünschten LDL‑C‑Ansprechen zu begrenzen.
Basierend auf Beobachtungen erniedrigter essentieller Fettsäure‑ und Vitamin‑E‑Spiegel in klinischen Studien sollten die Patienten während der gesamten Behandlung mit Lojuxta (siehe Abschnitt 4.4) täglich Nahrungsergänzungsmittel einnehmen, die 400 IE Vitamin E, etwa 200 mg Linolsäure, 110 mg Eicosapentaensäure (EPA), 210 mg Alpha‑Linolensäure (ALA) und 80 mg Docosahexaensäure (DHA) pro Tag enthalten.
Besondere Patientengruppen
Ältere Patienten
Es liegen begrenzte Erfahrungen mit Lomitapid bei Patienten über 65 Jahren vor. Daher ist bei diesen Patienten besondere Vorsicht geboten.
Da das empfohlene Dosierungsschema beinhaltet, mit der niedrigsten Dosis zu beginnen und die Dosis vorsichtig gemäß der individuellen Verträglichkeit des Patienten zu erhöhen, wird keine Dosisanpassung bei älteren Patienten empfohlen.
Eingeschränkte Leberfunktion
Lomitapid ist bei Patienten mit mittelschwerer oder schwerer Beeinträchtigung der Leber, einschließlich Patienten mit ungeklärten fortbestehenden anomalen Leberfunktionswerten, kontraindiziert (siehe Abschnitte 4.3 und 5.2).
Patienten mit leichter Beeinträchtigung der Leber (Child‑Pugh A) sollten eine Dosis von 40 mg pro Tag nicht überschreiten.
Eingeschränkte Nierenfunktion
Bei Patienten mit dialysepflichtiger terminaler Niereninsuffizienz sollte eine Dosis von 40 mg pro Tag nicht überschritten werden (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Lomitapid bei Kindern im Alter von < 18 Jahren ist nicht erwiesen; daher wird die Anwendung dieses Arzneimittels bei Kindern nicht empfohlen. Es liegen keine Daten vor.
Art der Anwendung
Zum Einnehmen.
Die Einnahme zusammen mit Nahrung kann die Exposition gegenüber Lomitapid erhöhen. Die Einnahme sollte auf nüchternen Magen erfolgen, mindestens 2 Stunden nach der Abendmahlzeit, da der Fettgehalt einer kürzlich eingenommenen Mahlzeit die gastrointestinale Verträglichkeit beeinträchtigen kann (siehe Abschnitt 4.4).
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Patienten mit mittelschwerer oder schwerer Beeinträchtigung der Leber sowie Patienten mit ungeklärten fortbestehenden anomalen Leberfunktionswerten (siehe Abschnitt 4.2).
Patienten mit bekannter signifikanter oder chronischer Darmerkrankung wie einer entzündlichen Darmerkrankung oder Malabsorption.
gleichzeitige Anwendung von > 40 mg Simvastatin (siehe Abschnitt 4.5).
gleichzeitige Anwendung von Lojuxta zusammen mit starken oder mittelstarken CYP3A4‑Hemmern (z. B. Azol‑Antimykotika wie Itraconazol, Fluconazol, Ketoconazol, Voriconazol oder Posaconazol; Makrolidantibiotika wie Erythromycin oder Clarithromycin; Antibiotika aus der Gruppe der Ketolide wie Telithromycin; HIV‑Protease‑Hemmer; die Kalziumkanalblocker Diltiazem und Verapamil sowie das Antiarrhythmikum Dronedaron [siehe Abschnitt 4.5]).
Schwangerschaft (siehe Abschnitt 4.6).
Leberenzymanomalien
Lomitapid kann zu Erhöhungen der Leberenzyme Alaninaminotransferase [ALT] und der Aspartataminotransferase [AST] sowie zu Steatosis hepatis führen (siehe Abschnitt 5.1). Es traten keine klinisch bedeutsamen Erhöhungen des Serum‑Bilirubins, der International Normalised Ratio (INR) oder der alkalischen Phosphatase als Begleiterscheinung, noch im Nachhinein auf. Inwieweit eine Lomitapid‑assoziierte Steatosis hepatis die Erhöhungen der Aminotransferasen fördert, ist unbekannt. Veränderungen der Leberenzymwerte können jederzeit während der Behandlung auftreten, treten allerdings meistens während der Dosiseskalation auf.
Obwohl keine Fälle von hepatischer Dysfunktion (erhöhte Aminotransferasen zusammen mit einem Anstieg der Bilirubinwerte oder der INR) oder Leberversagen berichtet wurden, bestehen Bedenken, dass Lomitapid eine Steatohepatitis induzieren könnte, die im Verlauf mehrerer Jahre zu einer Leberzirrhose fortschreiten könnte. Es ist aufgrund der Größe und Dauer der klinischen Studien zur Unterstützung der Sicherheit und Wirksamkeit von Lomitapid bei HoFH unwahrscheinlich, dass man dieses unerwünschte Ergebnis in diesen Studien festgestellt hätte.
Überwachung der Leberfunktionswerte
Vor Beginn der Behandlung mit Lojuxta sollten die Werte für ALT, AST, alkalische Phosphatase, Gesamtbilirubin, Gamma‑Glutamyltransferase (Gamma‑GT) und Serumalbumin gemessen werden. Das Arzneimittel ist bei Patienten mit mittelschwerer oder schwerer Beeinträchtigung der Leber und Patienten mit ungeklärten fortbestehenden anomalen Leberfunktionswerten kontraindiziert. Wenn die Ausgangsleberwerte Anomalien zeigen, sollte in Erwägung gezogen werden, die Behandlung mit dem Arzneimittel erst einzuleiten, nachdem eine angemessene Untersuchung durch einen Hepatologen durchgeführt wurde und die abnormalen Ausgangswerte geklärt wurden oder sich zurückgebildet haben.
Während des ersten Jahres sollten vor jeder Dosiserhöhung oder monatlich, je nachdem, was zuerst eintritt, Leberuntersuchungen durchgeführt werden (mindestens ALT und AST). Nach dem ersten Jahr sollten diese Untersuchungen mindestens alle 3 Monate und vor jeder Dosiserhöhung durchgeführt werden. Die Dosis von Lojuxta sollte gesenkt werden, wenn erhöhte Aminotransferasen festgestellt werden, und die Behandlung sollte bei persistierenden oder klinisch signifikanten Erhöhungen abgebrochen werden (siehe Tabelle 1).
Dosisänderung auf Grundlage erhöhter hepatischer Aminotransferasen
Tabelle 1 fasst die Empfehlungen für Dosisanpassungen und die Überwachung bei Patienten zusammen, die während der Therapie mit Lojuxta erhöhte Aminotransferasen entwickeln.

Wenn die Erhöhungen der Aminotransferasen mit klinischen Symptomen einer Leberschädigung (wie Übelkeit, Erbrechen, Abdominalschmerzen, Fieber, Gelbsucht, Lethargie, grippeartige Symptome), Anstieg der Bilirubinwerte auf ≥ 2x ULN oder einer aktiven Lebererkrankung einhergehen, sollte die Behandlung mit Lojuxta abgebrochen und der Patient zur weiteren Untersuchung an einen Hepatologen überwiesen werden.
Eine erneute Einleitung der Therapie kann in Erwägung gezogen werden, wenn angenommen wird, dass der Nutzen gegenüber den Risiken, die mit einer potenziellen Lebererkrankung assoziiert sind, überwiegt.
Steatosis hepatis und das Risiko einer progressiven Lebererkrankung
In Einklang mit dem Wirkmechanismus von Lomitapid zeigten die meisten behandelten Patienten Anstiege des Fettgehalts der Leber. In einer unverblindeten Phase‑III‑Studie entwickelten 18 von 23 Patienten mit HoFH eine Steatosis hepatis (Fettgehalt der Leber > 5,56 %), was anhand von Kernspinresonanzspektroskopie gemessen wurde (siehe Abschnitt 5.1). Der mediane absolute Anstieg des Leberfettgehalts betrug, gemessen anhand von Kernspinresonanzspektroskopie, 6 % nach sowohl 26 als auch 78 Behandlungswochen, ausgehend von einem Ausgangswert von 1 %. Steatosis hepatis ist ein Risikofaktor für eine progressive Lebererkrankung, einschließlich Steatohepatitis und Leberzirrhose. Die Langzeitfolgen einer Steatosis hepatis im Zusammenhang mit der Behandlung mit Lomitapid sind unbekannt. Klinische Daten deuten darauf hin, dass die Fettanreicherung der Leber nach Absetzen der Behandlung mit Lojuxta reversibel ist. Es ist jedoch nicht bekannt, ob histologische Folgeschäden zurückbleiben, insbesondere nach Langzeitanwendung.
Überwachung in Hinblick auf Nachweise einer progressiven Lebererkrankung
Ein reguläres Screening zur Feststellung einer Steatohepatitis/Fibrose sollte unter Verwendung der folgenden Bildgebungsverfahren und Biomarkermessungen vor Behandlungsbeginn sowie einmal jährlich durchgeführt werden:
Bildgebungsverfahren zur Darstellung der Gewebeelastizität, z. B. Fibroscan, Acoustic Radiation Force Impulse (ARFI) oder Magnetresonanz‑Elastografie (MR‑Elastografie)
Gamma‑GT und Serumalbumin zum Nachweis einer möglichen Leberschädigung
mindestens ein Marker aus jeder der folgenden Kategorien:
hochsensitives C‑reaktives Protein (hs‑CRP), Blutkörperchensenkungsrate (BKS), Fragment des CK 18, NashTest (Leberentzündung)
Enhanced Liver Fibrosis (ELF) Panel, Fibrometer, AST/ALT‑Quotient, Fib‑4‑Score, Fibrotest (Leberfibrose)
Die Durchführung und Interpretation dieser Tests sollten in Zusammenarbeit zwischen dem behandelnden Arzt und dem Hepatologen erfolgen. Bei Patienten mit Ergebnissen, die auf das Vorliegen einer Steatohepatitis oder Fibrose hindeuten, sollte eine Leberbiopsie in Erwägung gezogen werden.
Wenn ein Patient eine bioptisch nachgewiesene Steatohepatitis oder Fibrose aufweist, sollte das Nutzen‑Risiko‑Verhältnis neu beurteilt und die Behandlung, falls erforderlich, abgebrochen werden.
Dehydratation
Nach Markteinführung wurde über Dehydratation und Hospitalisierung bei Patienten, die mit Lomitapid behandelt wurden, berichtet. Patienten, die mit Lomitapid behandelt werden, sollten über die potenzielle Gefahr der Dehydratation in Bezug auf gastrointestinale Nebenwirkungen und über Vorsichtsmaßnahmen zur Vermeidung von Flüssigkeitsmangel informiert werden.
Gleichzeitige Anwendung von CYP3A4‑Hemmern
Lomitapid ist offenbar ein empfindliches CYP3A4‑Substrat. CYP3A4‑Hemmer erhöhen die Lomitapid‑Exposition, wobei starke Hemmer die Exposition um etwa das 27‑Fache erhöhen. Die gleichzeitige Anwendung von mittelstarken oder starken CYP3A4‑Hemmern zusammen mit Lojuxta ist kontraindiziert (siehe Abschnitt 4.3). In den klinischen Studien zu Lomitapid entwickelte ein Patient mit HoFH innerhalb weniger Tage nach Beginn der Behandlung mit dem starken CYP3A4‑Hemmer Clarithromycin deutlich erhöhte Aminotransferasespiegel (ALT 24x ULN, AST 13x ULN). Wenn die Behandlung mit mittelstarken oder starken CYP3A4‑Hemmern nicht vermeidbar ist, sollte die Einnahme von Lojuxta während dieser Behandlung unterbrochen werden.
Es ist zu erwarten, dass schwache CYP3A4‑Hemmer die Lomitapid‑Exposition erhöhen, wenn sie gleichzeitig eingenommen werden. Bei der Anwendung zusammen mit Atorvastatin sollte die Dosis von Lojuxta entweder in einem zeitlichen Abstand von 12 Stunden eingenommen werden oder um die Hälfte reduziert werden (siehe Abschnitt 4.2). Die Dosis von Lojuxta sollte in einem zeitlichen Abstand von 12 Stunden zu einem anderen schwachen CYP3A4‑Hemmer gegeben werden.
Gleichzeitige Anwendung von CYP3A4‑Induktoren
Bei Arzneimitteln, die CYP3A4 induzieren, kann davon ausgegangen werden, dass sie die Rate und das Ausmaß der Metabolisierung von Lomitapid erhöhen. CYP3A4‑Induktoren üben ihre Wirkung auf zeitabhängige Weise aus. Es kann mindestens 2 Wochen dauern, bis nach Beginn der Einnahme die maximale Wirkung erreicht wird. Im Gegenzug kann es bei deren Absetzen mindestens 2 Wochen dauern, bis die CYP3A4‑Induktion abnimmt.
Man kann davon ausgehen, dass durch die gleichzeitige Anwendung von CYP3A4‑Induktoren die Wirkung von Lomitapid gesenkt wird. Jeglicher Einfluss auf die Wirksamkeit ist wahrscheinlich variabel. Bei gleichzeitiger Anwendung von CYP3A4‑Induktoren (d. h. Aminoglutethimid, Nafcillin, nicht nukleosidische Reverse‑Transkriptase‑Inhibitoren, Phenobarbital, Rifampicin, Carbamazepin, Pioglitazon, Glukokortikoide, Modafinil und Phenytoin) zusammen mit Lojuxta sollte die Möglichkeit einer Wechselwirkung, welche die Wirksamkeit beeinträchtigt, berücksichtigt werden. Die Anwendung von Johanniskraut sollte während der Einnahme von Lojuxta vermieden werden.
Es wird empfohlen, während derartiger gleichzeitiger Anwendungen häufiger LDL‑C‑Beurteilungen durchzuführen und eine Erhöhung der Dosis von Lojuxta in Erwägung zu ziehen, um ein Aufrechterhalten des gewünschten Wirksamkeitsniveaus sicherzustellen, wenn der CYP3A4‑Induktor chronisch angewendet werden soll. Bei Absetzen eines CYP3A4‑Induktors sollte die Möglichkeit eines Anstiegs der Exposition berücksichtigt werden, und unter Umständen ist eine Senkung der Dosis von Lojuxta erforderlich.
Gleichzeitige Anwendung von HMG‑CoA‑Reduktasehemmern („Statine“)
Lomitapid erhöht die Plasmakonzentration von Statinen. Patienten, die Lojuxta zusätzlich zu einem Statin erhalten, sollten auf unerwünschte Ereignisse, die mit der Anwendung hoher Statin‑Dosen assoziiert sind, überwacht werden. Statine führen gelegentlich zu Myopathie. In seltenen Fällen manifestiert sich die Myopathie als Rhabdomyolyse mit oder ohne akutem Nierenversagen aufgrund von Myoglobinurie und kann einen tödlichen Ausgang haben. Alle Patienten, die Lomitapid zusätzlich zu einem Statin erhalten, sollten über das potenziell erhöhte Risiko einer Myopathie aufgeklärt werden und aufgefordert werden, ungeklärte Muskelschmerzen, ‑empfindlichkeit oder ‑schwäche umgehend mitzuteilen. Simvastatin‑Dosen von > 40 mg sollten nicht zusammen mit Lojuxta gegeben werden (siehe Abschnitt 4.3).
Grapefruitsaft
Grapefruitsaft muss während der Behandlung mit Lojuxta bei der Diät gemieden werden.
Risiko supratherapeutischer oder subtherapeutischer Antikoagulation mit Antikoagulanzien vom Cumarin‑Typ
Lomitapid erhöht die Plasmakonzentration von Warfarin. Erhöhungen der Dosis von Lojuxta können zu einer supratherapeutischen Antikoagulation und Senkungen der Dosis zu einer subtherapeutischen Antikoagulation führen. Schwierigkeiten, die INR zu kontrollieren, führten bei einem von 5 Patienten, die gleichzeitig Warfarin einnahmen, zu einem frühzeitigen Austritt aus der Phase‑III‑Studie. Bei Patienten, die Warfarin einnehmen, sollte der INR‑Wert regelmäßig überwacht werden, insbesondere nach Änderungen der Dosierung von Lojuxta. Die Warfarin‑Dosis sollte der klinischen Indikation entsprechend angepasst werden.
Alkoholkonsum
Alkohol kann die Fettspiegel in der Leber erhöhen und eine Leberschädigung verursachen oder verschlimmern. In der Phase‑III‑Studie berichteten 3 von 4 Patenten mit ALT‑Erhöhungen von > 5x ULN über Alkoholkonsum, der die im Prüfplan empfohlenen Grenzen überschritt. Der Konsum von Alkohol wird während der Behandlung mit Lomitapid nicht empfohlen.
Hepatotoxische Mittel
Vorsicht ist geboten, wenn Lojuxta mit anderen Arzneimitteln mit bekanntem hepatotoxischem Potenzial, etwa Isotretinoin, Amiodaron, Acetaminophen (> 4 g/Tag über ≥ 3 Tage/Woche), Methotrexat, Tetrazyklinen oder Tamoxifen, angewendet wird. Die Wirkung einer gleichzeitigen Anwendung von Lomitapid zusammen mit anderen hepatotoxischen Arzneimitteln ist nicht bekannt. Unter Umständen ist eine häufigere Überwachung der Leberwerte erforderlich.
Verringerte Resorption von fettlöslichen Vitaminen und Fettsäuren im Serum
Aufgrund seines Wirkmechanismus im Dünndarm kann Lomitapid die Resorption fettlöslicher Nährstoffe senken. In der Phase‑III‑Studie erhielten die Patienten täglich Nahrungsergänzungsmittel mit Vitamin E, Linolsäure, ALA, EPA und DHA. In dieser Studie sanken die medianen Baseline‑Serumspiegel von Vitamin E, ALA, Linolsäure, EPA, DHA und Arachidonsäure bis zur Woche 26 ab, hielten sich jedoch oberhalb der unteren Grenze des Referenzbereichs. Unerwünschte klinische Folgen dieser erniedrigten Spiegel waren bei einem bis zu 78 Wochen langen Behandlungszeitraum mit Lomitapid nicht zu beobachten. Mit Lojuxta behandelte Patienten sollten täglich Nahrungsergänzungsmittel, die 400 internationale Einheiten Vitamin E und etwa 200 mg Linolsäure, 210 mg ALA, 110 mg EPA und 80 mg DHA enthalten, einnehmen.
Maßnahmen zur Empfängnisverhütung bei Frauen im gebärfähigen Alter
Vor Beginn der Behandlung sollte bei Frauen im gebärfähigen Alter eine angemessene Beratung zu wirksamen Methoden zur Empfängnisverhütung stattfinden und eine wirksame Empfängnisverhütung eingeleitet werden. Patientinnen, die östrogenbasierte orale Kontrazeptiva einnehmen, sollten über den möglichen Verlust der Wirksamkeit durch Diarrhö und/oder Erbrechen aufgeklärt werden (siehe Abschnitt 4.5). Östrogenhaltige orale Kontrazeptiva sind schwache CYP3A4‑Hemmer (siehe Abschnitt 4.2).
Die Patientinnen sollten angewiesen werden, bei einer Schwangerschaft unverzüglich ihren Arzt aufzusuchen und die Einnahme von Lojuxta zu beenden (siehe Abschnitt 4.6.).
Sonstige Bestandteile mit bekannter Wirkung
Lactose
Lojuxta enthält Lactose. Patienten mit der seltenen hereditären Galactose‑Intoleranz, völligem Lactasemangel oder Glucose‑Galactose‑Malabsorption sollten dieses Arzneimittel nicht anwenden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Kapsel, d.h. es ist nahezu „natriumfrei“.
Wirkungen anderer Arzneimittel auf Lomitapid und sonstige Wechselwirkungen
Tabelle 2: Wechselwirkungen zwischen Lojuxta und anderen Arzneimitteln und sonstige Wechselwirkungen
Arzneimittel | Wirkungen auf die Lomitapidspiegel | Empfehlungen in Bezug auf die gleichzeitige Anwendung zusammen mit Lojuxta |
CYP3A4-Hemmer | Starke und mittelstarke Hemmer | Starke und mittelstarke Hemmer |
Schwache Hemmer | Schwache Hemmer | |
CYP3A4-Induktoren | Bei Arzneimitteln, die CYP3A4 induzieren, wird erwartet, dass sie die Rate und das Ausmaß der Metabolisierung von Lomitapid erhöhen. Folglich würde dies die Wirkung von Lomitapid senken. Jeglicher Einfluss auf die Wirksamkeit ist wahrscheinlich variabel. | Bei gleichzeitiger Anwendung von CYP3A4-Induktoren (d. h. Aminoglutethimid, Nafcillin, nicht nukleosidische Reverse-Transkriptase-Inhibitoren, Phenobarbital, Rifampicin, Carbamazepin, Pioglitazon, Johanniskraut, Glukokortikoide, Modafinil und Phenytoin) zusammen mit Lojuxta sollte die Möglichkeit einer Wechselwirkung, welche die Wirksamkeit beeinträchtigt, berücksichtigt werden. Es wird empfohlen, während derartiger gleichzeitiger Anwendungen häufiger LDL‑C-Beurteilungen durchzuführen und eine Erhöhung der Dosis von Lojuxta in Erwägung zu ziehen, um ein Aufrechterhalten des gewünschten Wirksamkeitsniveaus sicherzustellen, wenn der CYP3A4-Induktor chronisch angewendet werden soll. |
Gallensäurebinder | Lomitapid wurde nicht auf Wechselwirkungen mit Gallensäurebindern (Harze wie Colesevelam und Colestyramin) untersucht. | Da Gallensäurebinder die Resorption oraler Arzneimittel beeinflussen können, sollten Gallensäurebinder mindestens 4 Stunden vor oder mindestens 4 Stunden nach Lojuxta eingenommen werden. |
Wirkungen von Lomitapid auf andere Arzneimittel
HMG‑CoA‑Reduktase‑Hemmer („Statine“)
Lomitapid erhöht die Plasmakonzentration von Statinen. Wenn Lomitapid 60 mg im Steady‑State vor Simvastatin 40 mg angewendet wurde, erhöhten sich die AUC‑ und Cmax‑Werte der Simvastatinsäure um 68 % bzw. 57 %. Wenn Lomitapid 60 mg im Steady‑State vor Atorvastatin 20 mg angewendet wurde, erhöhten sich die AUC‑ und Cmax‑Werte der Atorvastatinsäure um 52 % bzw. 63 %. Wenn Lomitapid 60 mg im Steady‑State vor Rosuvastatin 20 mg angewendet wurde, erhöhte sich die Tmax von Rosuvastatin von 1 auf 4 Stunden, die AUC erhöhte sich um 32 % und die Cmax blieb unverändert. Das Risiko einer Myopathie im Zusammenhang mit Simvastatin ist dosisabhängig. Die Anwendung von Lojuxta ist bei Patienten, die mit hohen Simvastatin‑Dosen (> 40 mg) behandelt werden, kontraindiziert (siehe Abschnitte 4.3 und 4.4).
Antikoagulanzien vom Cumarin‑Typ
Wenn Lomitapid 60 mg im Steady‑State und 6 Tage nach Warfarin 10 mg angewendet wurde, erhöhte sich der INR‑Wert um das 1,26‑Fache. Die AUC‑Werte von R(+)‑Warfarin und S(‑)‑Warfarin erhöhten sich um 25 % bzw. 30 %. Die Cmax von R(+)‑Warfarin und S(‑)‑Warfarin erhöhten sich um 14 % bzw. 15 %. Bei Patienten die Antikoagulanzien vom Cumarin‑Typ (wie Warfarin) und Lojuxta gleichzeitig einnehmen, sollte vor Beginn der Behandlung mit Lojuxta der INR‑Wert bestimmt und regelmäßig überwacht sowie die Dosierung der Antikoagulanzien vom Cumarin‑Typ wie klinisch angezeigt angepasst werden (siehe Abschnitt 4.4).
Fenofibrat, Niacin und Ezetimib
Wenn Lomitapid im Steady‑State vor mikronisiertem Fenofibrat 145 mg, Niacin mit verlängerter Freisetzung 1 000 mg oder Ezetimib 10 mg angewendet wurde, wurden keine klinisch signifikanten Wirkungen auf die Exposition dieser Arzneimittel beobachtet. Bei gleichzeitiger Anwendung mit Lojuxta sind keine Dosisanpassungen erforderlich.
Orale Kontrazeptiva
Wenn Lomitapid 50 mg im Steady‑State zusammen mit östrogenbasierten oralen Kontrazeptiva angewendet wurde, wurden keine klinisch bedeutenden oder statistisch signifikanten Wirkungen auf die Pharmakokinetik der Bestandteile der oralen Kontrazeptiva (Ethinylestradiol und 17‑Deacetyl‑Norgestimat, dem Metaboliten von Norgestimat) beobachtet. Es ist nicht zu erwarten, dass Lomitapid die Wirksamkeit östrogenbasierter oraler Kontrazeptiva direkt beeinflusst. Allerdings können Diarrhö und/oder Erbrechen die Hormonresorption senken. In Fällen protrahierter oder schwerer Diarrhö und/oder Erbrechens über mehr als 2 Tage sollten nach Abklingen der Symptome 7 Tage lang zusätzliche Maßnahmen zur Empfängnisverhütung angewendet werden.
P‑gp‑Substrate
Lomitapid hemmt P‑gp in vitro und kann die Resorption von P‑gp‑Substraten erhöhen. Die gleichzeitige Anwendung von Lojuxta zusammen mit P‑gp‑Substraten (wie Aliskiren, Ambrisentan, Colchicin, Dabigatranetexilat, Digoxin, Everolimus, Fexofenadin, Imatinib, Lapatinib, Maraviroc, Nilotinib, Posaconazol, Ranolazin, Saxagliptin, Sirolimus, Sitagliptin, Talinolol, Tolvaptan oder Topotecan) kann die Resorption der P‑gp‑Substrate erhöhen. Eine Senkung der Dosis des P‑gp‑Substrats sollte bei gleichzeitiger Anwendung zusammen mit Lojuxta in Erwägung gezogen werden.
In‑vitro‑Beurteilung von Wechselwirkungen
Lomitapid hemmt CYP3A4. Lomitapid induziert nicht die CYPs 1A2, 3A4 und 2B6 und hemmt auch nicht die CYPs 1A2, 2B6, 2C9, 2C19, 2D6 oder 2E1. Lomitapid ist kein P‑gp‑Substrat, hemmt jedoch P‑gp. Lomitapid hemmt nicht das Breast Cancer Resistance Protein (BCRP).
Anwendung bei Frauen im gebärfähigen Alter
Vor Beginn der Behandlung von Frauen im gebärfähigen Alter sollte das Nichtbestehen einer Schwangerschaft bestätigt, eine angemessene Beratung zu wirksamen Methoden zur Empfängnisverhütung durchgeführt und eine wirksame Empfängnisverhütung eingeleitet werden. Patientinnen, die östrogenbasierte orale Kontrazeptiva einnehmen, sollten über den möglichen Verlust der Wirksamkeit durch Diarrhö und/oder Erbrechen aufgeklärt werden. Bis zum Abklingen der Symptome sollten zusätzliche Maßnahmen zur Empfängnisverhütung angewendet werden (siehe Abschnitt 4.5).
Schwangerschaft
Lojuxta ist während der Schwangerschaft kontraindiziert. Es liegen keine zuverlässigen Daten für seine Anwendung bei Schwangeren vor. Tierexperimentelle Studien haben eine Entwicklungstoxizität aufgezeigt (Teratogenität, Embryotoxizität, siehe Abschnitt 5.3). Das potenzielle Risiko für den Menschen ist nicht bekannt.
Stillzeit
Es ist nicht bekannt, ob Lomitapid in die Muttermilch ausgeschieden wird. Aufgrund des Potenzials für schädliche Wirkungen beruhend auf den Befunden aus tierexperimentellen Studien mit Lomitapid (siehe Abschnitt 5.3) muss eine Entscheidung darüber getroffen werden, ob entweder das Stillen oder die Einnahme des Arzneimittels zu unterbrechen ist, wobei die Wichtigkeit der Einnahme des Arzneimittels für die Mutter berücksichtigt werden sollte.
Fertilität
Bei männlichen und weiblichen Ratten, denen Lomitapid in systemischen Expositionen (AUC) gegeben wurde, die nach Schätzungen 4‑ bis 5‑fach höher als beim Menschen unter der maximalen empfohlenen humantherapeutischen Dosis sind, wurden keine schädlichen Wirkungen auf die Fertilität beobachtet (siehe Abschnitt 5.3).
Lojuxta hat einen geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die schwersten Nebenwirkungen während der Behandlung waren Anomalien der Leber‑Aminotransferasen (siehe Abschnitt 4.4).
Die häufigsten Nebenwirkungen waren gastrointestinale Effekte. Gastrointestinale Nebenwirkungen wurden von 27 (93 %) der 29 Patienten in der klinischen Phase‑III‑Studie berichtet. Diarrhö trat bei 79 % der Patienten auf, Übelkeit bei 65 %, Dyspepsie bei 38 % und Erbrechen bei 34 %. Sonstige Reaktionen, die von mindestens 20 % der Patienten berichtet wurden, waren Abdominalschmerzen, abdominale Beschwerden, abdominelle Distension, Obstipation und Flatulenz. Gastrointestinale Nebenwirkungen traten häufiger während der Phase der Dosiserhöhung in der Studie auf und gingen zurück, nachdem bei den Patienten die höchste vertragene Dosis von Lomitapid erreicht wurde.
Gastrointestinale Nebenwirkungen von hoher Intensität wurden von 6 (21 %) der 29 Patienten in der klinischen Phase‑III‑Studie berichtet, wobei Diarrhö (4 Patienten, 14 %), Erbrechen (3 Patienten, 10 %) sowie Abdominalschmerzen, abdominelle Distension und/oder abdominale Beschwerden (2 Patienten, 7 %) am häufigsten waren. Gastrointestinale Reaktionen gehörten bei 4 (14 %) der Patienten zu den Gründen für einen frühzeitigen Austritt aus der Studie.
Die am häufigsten berichteten Nebenwirkungen hoher Intensität waren Diarrhö (4 Probanden, 14 %), Erbrechen (3 Patienten, 10 %) sowie abdominelle Distension und erhöhte ALT‑Werte (jeweils 2 Probanden, 7 %).
Tabellarische Auflistung der Nebenwirkungen
Die Nebenwirkungen sind im Folgenden gemäß Systemorganklasse (SOC) und nach Häufigkeit aufgeführt, wobei die häufigsten Nebenwirkungen zuerst aufgeführt sind. Die Häufigkeit der Nebenwirkungen ist wie folgt definiert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
In Tabelle 3 sind alle Nebenwirkungen aufgelistet, die unter den 35 in der Phase‑II‑Studie UP1001 und in der Phase‑III‑Studie UP1002/AEGR‑733‑005 oder in der Erweiterungsstudie AEGR‑733‑012 behandelten Patienten berichtet wurden.
Tabelle 3: Häufigkeit von Nebenwirkung bei Patienten mit HoFH
Systemorganklasse | Häufigkeit | Nebenwirkung |
Infektionen und parasitäre Erkrankungen | Häufig | Gastroenteritis |
Stoffwechsel- und Ernährungsstörungen | Sehr häufig | Verminderter Appetit |
Nicht bekannt | Dehydratation | |
Erkrankungen des Nervensystems | Häufig | Schwindelgefühl |
Erkrankungen des Gastrointestinaltrakts | Sehr häufig | Diarrhö |
Häufig | Gastritis | |
Leber- und Gallenerkrankungen | Häufig | Steatosis hepatis |
Erkrankungen der Haut und des Unterhautzellgewebes | Häufig | Ekchymose |
Nicht bekannnt | Alopezie | |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | Nicht bekannt | Myalgie |
Allgemeine Erkrankungen und Beschwerden am Anwendungsort | Häufig | Ermüdung |
Untersuchungen | Sehr häufig | Alaninaminotransferase erhöht |
Häufig | International Normalised Ratio erhöht |
In Tabelle 4 sind alle Nebenwirkungen aufgelistet, die bei Probanden berichtet wurden, die in den Phase‑II‑Studien an Probanden mit erhöhtem LDL‑C (N = 462) Lomitapid als Monotherapie (N = 291) erhalten hatten.
Tabelle 4: Häufigkeit der Nebenwirkungen bei Patienten mit erhöhtem LDL‑C
Systemorganklasse | Häufigkeit | Nebenwirkung |
Infektionen und parasitäre Erkrankungen | Gelegentlich | Gastroenteritis |
Erkrankungen des Blutes und des Lymphsystems | Gelegentlich | Anämie |
Stoffwechsel- und Ernährungsstörungen | Häufig | Verminderter Appetit |
Gelegentlich | Dehydratation | |
Erkrankungen des Nervensystems | Gelegentlich | Parästhesie |
Augenerkrankungen | Gelegentlich | Schwellung des Auges |
Erkrankungen des Ohrs und des Labyrinths | Gelegentlich | Vertigo |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | Gelegentlich | Läsion des Pharynx |
Erkrankungen des Gastrointestinaltrakts | Sehr häufig | Diarrhö |
Häufig | Schmerzen im Oberbauch | |
Gelegentlich | Mundtrockenheit | |
Leber- und Gallenerkrankungen | Gelegentlich | Hepatomegalie |
Erkrankungen der Haut und des Unterhautzellgewebes | Gelegentlich | Blasen |
Skelettmuskulatur, Bindegewebs- und Knochenerkrankungen | Häufig | Muskelspasmen |
Gelegentlich | Arthralgie | |
Erkrankungen der Nieren und Harnwege | Gelegentlich | Hämaturie |
Allgemeine Erkrankungen und Beschwerden am Anwendungsort | Häufig | Ermüdung |
Gelegentlich | Brustschmerzen | |
Untersuchungen | Häufig | Alaninaminotransferase erhöht |
Gelegentlich | Gewicht erniedrigt |
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen‑Risiko‑Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das
Bundesinstitut für Arzneimittel und Medizinprodukte
Abt. Pharmakovigilanz
Kurt-Georg-Kiesinger-Allee 3
D-53175 Bonn
Website: http://www.bfarm.de
anzuzeigen.
Es gibt keine spezielle Therapie bei einer Überdosierung. Im Falle einer Überdosierung ist der Patient symptomatisch zu behandeln, bei Bedarf sind unterstützende Maßnahmen einzuleiten. Die Leberwerte sollten überwacht werden. Eine Hämodialyse ist wenig zielführend, da Lomitapid stark proteingebunden ist.
Bei Nagetieren wurden orale Einzeldosen von Lomitapid, die ≥ 600 Mal höher als die empfohlene Höchstdosis beim Menschen (1 mg/kg) waren, gut vertragen. Die in klinischen Studien bei Probanden angewendete Höchstdosis betrug 200 mg als Einzeldosis. Es traten keine Nebenwirkungen auf.
Pharmakotherapeutische Gruppe: Mittel, die den Lipidstoffwechsel beeinflussen, andere Mittel, die den Lipidstoffwechsel beeinflussen. ATC‑Code: C10AX12.
Wirkmechanismus
Lomitapid ist ein selektiver Hemmer des mikrosomalen Transfer‑Proteins (MTP), ein intrazelluläres Lipid‑Transfer‑Protein, das im Lumen des endoplasmatischen Retikulums vorkommt und für die Bindung und den Transport einzelner Lipidmoleküle zwischen Membranen verantwortlich ist. MTP spielt eine wichtige Rolle bei der Zusammensetzung Apo‑B‑haltiger Lipoproteine in der Leber und im Darm. Die Hemmung des MTP senkt die Sekretion von Lipoproteinen und die zirkulierenden Konzentrationen von Lipiden, die von Lipoproteinen transportiert werden, einschließlich Cholesterin und Triglyzeriden.
Klinische Wirksamkeit und Sicherheit
In einer einarmigen, unverblindeten Studie (UP1002/AEGR‑733‑005) wurden die Wirksamkeit und Sicherheit von Lomitapid bei Anwendung zusammen mit einer fettarmen Diät und anderen lipidsenkenden Therapien bei Erwachsenen mit HoFH bewertet. Die Patienten wurden angewiesen, ab 6 Wochen vor Beginn der Behandlung bis einschließlich mindestens Woche 26 eine fettarme Diät (< 20 % der Kalorien von Fett stammend) und ihre bei Eintritt in die Studie bestehenden lipidsenkenden Therapien, einschließlich ggf. Apherese, einzuhalten. Die Lomitapid‑Dosis wurde von 5 mg schrittweise auf eine individuell bestimmte höchste vertragene Dosis von bis zu 60 mg erhöht. Nach Woche 26 führten die Patienten die Einnahme von Lomitapid zur Bestimmung der Wirkungen in einer Langzeitbehandlung fort, durften jedoch die vorbestehenden lipidsenkenden Therapien ändern. Die Studie umfasste insgesamt 78 Behandlungswochen.
29 Patienten wurden in die Studie aufgenommen, von denen 23 Woche 78 abschlossen. Es wurden 16 Männer (55 %) und 13 Frauen (45 %) mit einem mittleren Alter von 30,7 Jahren (Bereich: 18‑55 Jahre) eingeschlossen. Die mittlere Lomitapid‑Dosis betrug 45 mg in Woche 26 und 40 mg in Woche 78. In Woche 26 betrug die mittlere prozentuale LDL‑C‑Veränderung im Vergleich zu den LDL‑C‑Ausgangswerten in der Intent‑to‑Treat‑Population (ITT‑Population) 40 % (p< 0,001). Die mittlere prozentuale Veränderung ausgehend von der Baseline bis einschließlich Woche 26 anhand des letzten beobachteten Werts (LOCF) für jede Beurteilung ist in Abbildung 1 aufgeführt.
Abbildung 1: Mittlere prozentuale LDL‑C‑Veränderung ab Baseline in der Hauptwirksamkeitsstudie UP1002/AEGR‑733‑005 bis einschließlich Woche 26 (der primäre Endpunkt) anhand des LOCF für jede Beurteilung (N = 29)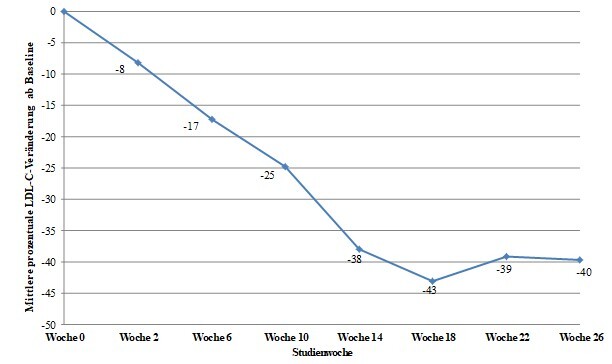
Veränderungen bei den Lipiden und Lipoproteinen bis einschließlich Woche 26 und Woche 78 der Lomitapid‑Behandlung sind in Tabelle 5 aufgeführt.
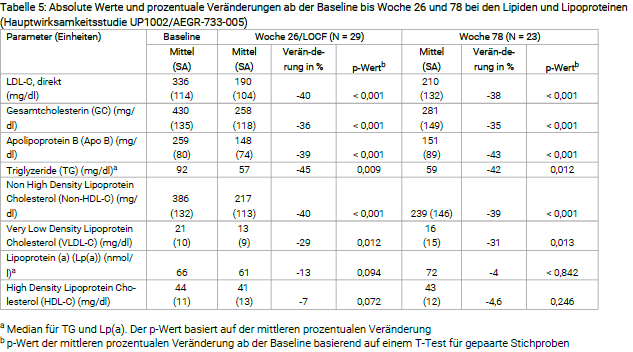
Sowohl in Woche 26 als auch Woche 78 zeigten sich signifikante Senkungen von LDL‑C, GC, Apo B, TG, Non‑HDL‑C und VLDL‑C. Die Veränderungen des HDL‑C zeigten in Woche 26 eine abfallende Tendenz, und bis Woche 78 stiegen die HDL‑C‑Spiegel wieder auf die Ausgangswerte an.
Die Wirkung von Lojuxta auf die kardiovaskuläre Morbidität und Mortalität wurde nicht bestimmt.
Bei Beginn der Behandlung erhielten 93 % ein Statin, 76 % Ezetimib, 10 % Niacin, 3 % einen Gallensäurebinder und 62 % Apherese. 15 von 23 Patienten (65 %) reduzierten bis Woche 78 ihre lipidsenkende Behandlung, einschließlich geplanter und ungeplanter Dosisreduzierungen/Unterbrechungen. Bei 3 von 13 mit Apherese behandelten Patienten wurde die Apherese in Woche 26 abgesetzt und bei 3 Patienten die Häufigkeit unter Aufrechterhaltung niedriger LDL‑C‑Spiegel bis einschließlich Woche 78 gesenkt. Der klinische Nutzen der Senkungen der vorbestehenden lipidsenkenden Therapien, einschließlich Apherese, ist nicht gesichert.
Von den 23 Patienten, die Woche 78 abschlossen, wiesen 19 (83 %) LDL‑C‑Senkungen von ≥ 25 % auf, wobei zu diesem Zeitpunkt 8 (35 %) LDL‑C‑Spiegel von < 100 mg/dl hatten und 1 einen LDL‑C‑Spiegel von < 70 mg/dl hatte.
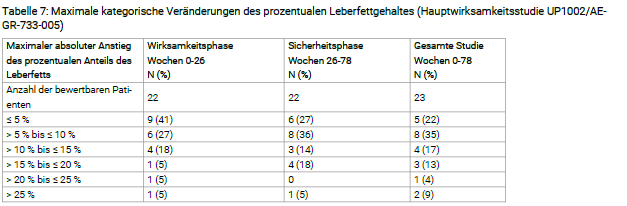
In dieser Studie zeigten 10 Patienten Erhöhungen der AST‑ und/oder ALT‑Werte von > 3 x ULN (siehe Tabelle 6).
Tabelle 6: Höchste Ergebnisse der Leberfunktionstests nach der ersten Dosis (Hauptwirksamkeitsstudie UP1002/AEGR‑733‑005)
Parameter/Anomalie | N (%) |
ALT | |
Anzahl der Patienten mit Beurteilungen | 29 |
> 3 bis ≤ 5 x ULN | 6 (20,7) |
> 5 bis ≤ 10 x ULN | 3 (10,3) |
> 10 bis ≤ 20 x ULN | 1 (3,4) |
> 20 x ULN | 0 |
AST | |
Anzahl der Patienten mit Beurteilungen | 29 |
> 3 bis ≤ 5 x ULN | 5 (17,2) |
> 5 bis ≤ 10 x ULN | 1 (3,4) |
> 10 bis ≤ 20 x ULN | 0 |
> 20 x ULN | 0 |
Erhöhungen der ALT‑ und/oder AST‑Werte von > 5 x ULN wurden mit einer Dosisreduktion oder einem vorübergehenden Absetzen von Lomitapid behandelt. Alle Patienten konnten die Behandlung mit dem Prüfpräparat fortsetzen. Es wurden keine klinisch bedeutenden Erhöhungen beim Gesamtbilirubin oder der alkalischen Phosphatase beobachtet. Der Fettgehalt der Leber wurde bei allen geeigneten Patienten während der klinischen Studie prospektiv mithilfe von Kernspinresonanzspektroskopie gemessen (Tabelle 7). Daten von Einzelpersonen, bei denen nach Beenden der Behandlung mit Lomitapid wiederholte Messungen durchgeführt wurden, zeigen, dass die Fettanhäufung in der Leber reversibel ist. Es ist jedoch nicht bekannt, ob histologische Folgeschäden zurückbleiben.
Die Europäische Arzneimittel‑Agentur hat für Lojuxta eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in HoFH gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Dieses Arzneimittel wurde unter „Außergewöhnlichen Umständen“ zugelassen. Das bedeutet, dass es aufgrund der Seltenheit der Erkrankung nicht möglich war vollständige Informationen zu diesem Arzneimittel zu erhalten.
Die Europäische Arzneimittel‑Agentur wird alle neuen Informationen, die verfügbar werden, jährlich bewerten, und falls erforderlich, wird die Zusammenfassung der Merkmale des Arzneimittels aktualisiert werden.
Resorption
Die absolute orale Bioverfügbarkeit von Lomitapid beträgt 7 %. Die Resorption ist nicht durch die Passage des Wirkstoffs durch die Darmschranke begrenzt, wird aber vorherrschend von einem ausgeprägten First‑Pass‑Effekt beeinflusst. Die Spitzen‑Plasmakonzentrationen von Lomitapid wurden 4‑8 Stunden nach oraler Dosis erzielt. Die Pharmakokinetik von Lomitapid ist bei oralen Einzeldosen im therapeutischen Bereich in etwa dosisproportional. Dosen von mehr als 60 mg weisen auf eine Tendenz in Richtung Nichtlinearität hin und werden nicht empfohlen.
Nach mehrfachen Dosen stiegen die Cmax‑ und AUC‑Werte in etwa proportional zur Lomitapid‑Dosis an. Erhöhte Cmax‑ und AUC‑Werte zeigten sich sowohl nach einer fettreichen Mahlzeit (77 % bzw. 58 %) als auch nach einer fettarmen Mahlzeit (70 % bzw. 28 %). Die Akkumulation von Lomitapid im Plasma stimmte mit den Voraussagen für die Akkumulation nach einer Einzeldosis nach einmal täglich gegebenen Dosen von mehr als 25 mg über bis zu 4 Wochen überein. Die interindividuelle Variabilität bei der AUC von Lomitapid betrug etwa 50 %.
Im Steady‑State betrug die Akkumulation von Lomitapid 2,7 bei 25 mg und 3,9 bei 50 mg.
Verteilung
Nach intravenöser Verabreichung war das Verteilungsvolumen von Lomitapid trotz hochgradiger (> 99,8 %) Bindung an das Plasmaprotein hoch (Mittel = 1 200 Liter). In tierexperimentellen Studien war Lomitapid in der Leber hoch konzentriert (200‑fach).
Biotransformation
Lomitapid wird weitgehend metabolisiert, vorwiegend durch CYP3A4. Die CYP‑Isoformen 2E1, 1A2, 2B6, 2C8 und 2C19 sind in einem geringeren Ausmaß beteiligt. Die Isoformen 2D6 und 2C9 sind nicht am Metabolismus von Lomitapid beteiligt.
Elimination
Nach Gabe einer radioaktiv markierten Dosis einer oralen Lösung an gesunde Probanden wurden 93 % der angewendeten Dosis im Urin und in den Faeces gefunden. Etwa 33 % der Radioaktivität wurden als Metaboliten über den Urin ausgeschieden. Der Rest wurde über die Faeces ausgeschieden, überwiegend als oxidierte Metaboliten. Die Eliminationshalbwertszeit von Lomitapid betrug etwa 29 Stunden.
Besondere Patientengruppen
Daten aus der zulassungsrelevanten klinischen Studie wurden hinsichtlich des Einflusses potenzieller Kovariaten auf die Lomitapid‑Exposition analysiert. Von den untersuchten Parametern (ethnische Zugehörigkeit, Body‑Mass‑Index (BMI), Geschlecht, Gewicht, Alter) konnte nur der BMI als potenzielle Kovariate klassifiziert werden.
Alter und Geschlecht
Alter (18‑64 Jahre) oder Geschlecht zeigten keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Lomitapid. Lomitapid wurde bei Patienten ab 65 Jahren nicht untersucht.
Ethnische Zugehörigkeit
Bei kaukasischen oder lateinamerikanischen Patienten ist keine Dosisanpassung erforderlich. Es liegen unzureichende Informationen vor, um zu bestimmen, ob Lojuxta bei anderen ethnischen Zugehörigkeiten eine Dosisanpassung erfordert. Da das Arzneimittel allerdings schrittweise entsprechend der individuellen Sicherheit und Verträglichkeit des Patienten erhöht wird, wird keine Dosisanpassung basierend auf der ethnischen Zugehörigkeit empfohlen.
Niereninsuffizienz
In der Population mit Niereninsuffizienz wurde Lomitapid nur bei Patienten mit terminaler Niereninsuffizienz (end‑stage renal disease, ESRD) untersucht. Eine pharmakokinetische Studie an Patienten mit dialysepflichtiger terminaler Niereninsuffizienz zeigte im Vergleich zu entsprechenden gesunden Kontrollen einen Anstieg der mittleren Plasmakonzentration von Lomitapid um 36 %. Die terminale Halbwertszeit von Lomitapid wurde nicht beeinflusst.
Leberinsuffizienz
Es wurde eine unverblindete Studie mit Einzeldosen durchgeführt, um die Pharmakokinetik von 60 mg Lomitapid bei gesunden Probanden mit normaler Leberfunktion im Vergleich zu Patienten mit leichter (Child‑Pugh A) und mittelschwerer (Child‑Pugh B) Beeinträchtigung der Leber zu bewerten. Im Vergleich zu gesunden Probanden waren die AUC‑ und Cmax‑Werte von Lomitapid bei Patienten mit mittelschwerer Beeinträchtigung der Leber um 164 % bzw. 361 % erhöht. Bei Patienten mit leichter Beeinträchtigung der Leber waren die AUC‑ und Cmax‑Werte von Lomitapid im Vergleich zu gesunden Probanden um 47 % bzw. 4 % erhöht. Lojuxta wurde bei Patienten mit schwerer Beeinträchtigung der Leber (Child‑Pugh‑Score 10‑15) nicht untersucht.
Kinder und Jugendliche
Lomitapid wurde bei Kindern unter 18 Jahren nicht untersucht.
In Studien zur Toxizität nach wiederholter oraler Gabe an Nagetieren und Hunden waren die wirkstoffbedingten Hauptbefunde eine Lipidakkumulation im Dünndarm und/oder in der Leber im Zusammenhang mit Senkungen der Cholesterin‑ und/oder Triglyzeridspiegel im Serum. Diese Veränderungen sind eine Folge des Wirkmechanismus von Lomitapid. Veränderungen in Bezug auf die Leber in Studien zur Toxizität nach wiederholter Gabe bei Ratten und Hunden umfassten erhöhte Aminotransferasen im Serum, subakute Entzündungen (nur Ratten) und Einzelzellnekrosen. In einer 1‑Jahres‑Studie mit wiederholter Gabe an Hunden zeigten sich keine mikroskopischen Veränderungen in der Leber, obwohl die AST‑Werte im Serum bei weiblichen Tieren minimal erhöht waren.
Bei Nagetieren wurden pulmonale Histiozytosen beobachtet. Bei Hunden wurden erniedrigte Erythrozytenparameter sowie Poikilozytosen und/oder Anisozytosen beobachtet. Eine testikuläre Toxizität wurde bei Hunden in einer 6‑Monats‑Studie bei Dosen, die dem 205‑Fachen der menschlichen Exposition (AUC) bei 60 mg entsprachen, beobachtet. In einer 1‑Jahres‑Studie an Hunden wurden bei Dosen, die dem 64‑Fachen der menschlichen Exposition bei 60 mg entsprachen, keine schädlichen Wirkungen auf die Hoden beobachtet.
In einer Studie zur Kanzerogenität mit Gabe im Futter an Mäusen wurde Lomitapid bis zu 104 Wochen in Dosen zwischen 0,3 bis 45 mg/kg/Tag gegeben. Es bestanden statistisch signifikante Anstiege der Inzidenz von Leberadenomen und Karzinomen bei Dosen ≥ 1,5 mg/kg/Tag bei männlichen Tieren (≥ 2‑Fache der menschlichen Exposition bei 60 mg basierend auf der AUC) und ≥ 7,5 mg/kg/Tag bei weiblichen Tieren (≥ 9‑Fache der menschlichen Exposition bei 60 mg basierend auf der AUC). Die Inzidenz von Dünndarmkarzinomen und/oder Adenomen und Karzinomen in Kombination (seltene Tumore bei Mäusen) war bei Dosen ≥ 15 mg/kg/Tag bei männlichen Tieren (≥ 26‑Fache der menschlichen Exposition bei 60 mg täglich basierend auf der AUC) und bei Dosen von 15 mg/kg/Tag bei weiblichen Tieren (≥ 22‑Fache der menschlichen Exposition bei 60 mg basierend auf der AUC) signifikant erhöht.
In einer oralen Kanzerogenitätsstudie an Ratten wurde Lomitapid bis zu 99 Wochen männlichen Tieren in Dosen von bis zu 7,5 mg/kg/Tag und weiblichen Tieren in Dosen von bis zu 2,0 mg/kg/Tag gegeben. Fokale Leberfibrosen wurden bei männlichen und weiblichen Tieren beobachtet. Zystische Degenerationen der Leber wurden nur bei männlichen Tieren beobachtet. Bei männlichen Tieren unter hohen Dosen wurde bei einer Exposition, die basierend auf der AUC dem 6‑Fachen der menschlichen Exposition bei 60 mg entspricht, eine erhöhte Inzidenz von Azinuszelladenomen des Pankreas beobachtet.
Lomitapid erwies sich in einer Reihe von In‑vitro‑ und In‑vivo‑Studien weder als mutagen noch genotoxisch.
Lomitapid hatte bei weiblichen Ratten in Dosen von bis zu 1 mg/kg und bei männlichen Ratten in Dosen von bis zu 5 mg/kg keine Wirkung auf die Fortpflanzungsfähigkeit. Man schätzt, dass die systemische Exposition gegenüber Lomitapid bei diesen Dosen dem 4‑Fachen (weibliche Tiere) bzw. 5‑Fachen (männliche Tiere) der menschlichen Exposition bei 60 mg entspricht.
Lomitapid erwies sich als teratogen bei Ratten bei Abwesenheit maternaler Toxizität bei einer Exposition (AUC), die schätzungsweise dem 2‑Fachen der menschlichen Exposition bei 60 mg entspricht. Es gab keinen Beleg für eine embryofötale Toxizität in Kaninchen bei Dosen, die basierend auf der Körperoberfläche dem 3‑Fachen der maximalen empfohlenen humantherapeutischen Dosis (maximum recommended human dose, MRHD) von 60 mg entsprechen. Eine embryofetale Toxizität wurde bei Kaninchen bei Abwesenheit maternaler Toxizität mit Dosen beobachtet, die dem ≥ 6,5‑Fachen der MRHD entsprechen. Bei Frettchen war Lomitapid in Dosen, die dem < 1‑Fachen der MRHD entsprechen, sowohl maternal toxisch als auch teratogen.
Kapselinhalt:
Vorverkleisterte Stärke (Mais)
Carboxymethylstärke‑Natrium (Typ A) (Ph.Eur.)
Mikrokristalline Cellulose
Lactose‑Monohydrat
Hochdisperses Siliciumdioxid
Magnesiumstearat (Ph.Eur.)
Kapselhülle:
Lojuxta 5 mg, 10 mg Hartkapseln
Gelatine
Titandioxid (E171)
Eisen(III)‑oxid (E172)
Lojuxta 20 mg Hartkapseln
Gelatine
Titandioxid (E171)
Lojuxta 30 mg Hartkapseln
Gelatine
Titandioxid (E171)
Eisen(III)‑oxid (E172)
Eisen(III)‑hydroxid‑oxid × H2O (E172)
Lojuxta 40 mg, 60 mg Hartkapseln
Gelatine
Titandioxid (E171)
Eisen(III)‑hydroxid‑oxid × H2O (E172)
Druckfarbe:
Schellack
Eisen(II,III)‑oxid (E172)
Propylenglycol
Nicht zutreffend.
3 Jahre.
Nicht über 30 °C lagern.
Die Flasche fest verschlossen halten, um den Inhalt vor Feuchtigkeit zu schützen.
Flasche aus Polyethylen hoher Dichte (HDPE) mit einer Induktionsversiegelung aus Polyester/Aluminiumfolie/Karton und einem Polypropylen‑Schraubdeckel.
Die Packungsgrößen sind:
28 Kapseln
Keine besonderen Anforderungen.
Chiesi Farmaceutici S.p.A.
Via Palermo 26/A
43122 Parma
Italien
EU/1/13/851/001 – Lojuxta 5 mg Hartkapseln
EU/1/13/851/002 – Lojuxta 10 mg Hartkapseln
EU/1/13/851/003 – Lojuxta 20 mg Hartkapseln
EU/1/13/851/004 – Lojuxta 30 mg Hartkapseln
EU/1/13/851/005 – Lojuxta 40 mg Hartkapseln
EU/1/13/851/006 – Lojuxta 60 mg Hartkapseln
Datum der Erteilung der Zulassung: 31. Juli 2013
Datum der letzten Verlängerung der Zulassung: 26. Mai 2023
August 2024
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel‑Agentur https://www.ema.europa.eu verfügbar.
---------------------------------------------------------------------------------------------------------------------------
Verschreibungspflichtig
Chiesi GmbH
Gasstraße 6
22761 Hamburg
Telefon: 040 89724-0
Telefax: 040 89724-212
E-Mail: info.de@chiesi.com