▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung im Fertigpen
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung in einer Fertigspritze
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung im Fertigpen
Jeder Einweg-Fertigpen enthält nach der Rekonstitution 30 mg Nemolizumab pro 0,49-ml-Dosis.
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung in einer Fertigspritze
Jede Einweg-Fertigspritze enthält nach der Rekonstitution 30 mg Nemolizumab pro 0,49-ml-Dosis.
Nemolizumab, ein humanisierter monoklonaler modifizierter Immunglobulin-G (IgG)-Antikörper, wird mittels rekombinanter DNA-Technologie aus Ovarialzellen des chinesischen Hamsters gewonnen.
Vollständige Auflistung der sonstigen Bestandteile siehe Abschnitt 6.1.
Pulver und Lösungsmittel zur Herstellung einer Injektionslösung
Pulver zur Herstellung einer Injektionslösung: lyophilisiertes weißes Pulver.
Lösungsmittel zur Herstellung einer Injektionslösung: Eine klare, farblose Lösung.
Atopische Dermatitis (AD)
Nemluvio wird zur Behandlung von mittelschwerer bis schwerer atopischer Dermatitis bei Patienten ab 12 Jahren angewendet, die für eine systemische Therapie in Betracht kommen.
Prurigo nodularis (PN)
Nemluvio wird zur Behandlung von Erwachsenen mit mittelschwerer bis schwerer Prurigo nodularis angewendet, die für eine systemische Therapie in Betracht kommen.
Die Behandlung mit Nemolizumab sollte von Ärzten eingeleitet und überwacht werden, die in der Diagnose und Behandlung von Erkrankungen erfahren sind, für die Nemolizumab angewendet wird.
Dosierung
Atopische Dermatitis (AD)
Die empfohlene Dosis ist:
Eine Anfangsdosis von 60 mg (zwei 30-mg-Injektionen), gefolgt von 30 mg alle 4 Wochen (Q4W)
Nach 16 Behandlungswochen beträgt die empfohlene Erhaltungsdosis für Patienten, die ein klinisches Ansprechen erreichen, 30 mg alle 8 Wochen (Q8W).
Nemolizumab kann mit oder ohne topische Corticosteroide (TCS) angewendet werden. Topische Calcineurin-Inhibitoren (TCI) können angewendet werden, sollten aber nur auf die Problemzonen wie Gesicht, Hals, intertriginöse Bereiche und Genitalbereiche beschränkt bleiben. Jede Anwendung topischer Therapien sollte ausgeschlichen und anschließend abgesetzt werden, wenn eine ausreichende Besserung der Erkrankung erreicht wurde.
Bei Patienten, die nach 16 Wochen Behandlung der atopischen Dermatitis kein Ansprechen gezeigt haben, sollte ein Abbruch der Behandlung in Betracht gezogen werden. Bei Patienten mit einem anfänglichen partiellen Ansprechen kann sich der Zustand durch eine fortgesetzte Behandlung über 16 Wochen hinaus weiter verbessern.
Sobald ein klinisches Ansprechen erreicht ist, beträgt die empfohlene Erhaltungsdosis von Nemolizumab 30 mg alle 8 Wochen.
Prurigo nodularis (PN)
Die empfohlene Dosis für Patienten mit einem Gewicht von < 90 kg ist eine Anfangsdosis von 60 mg (zwei 30-mg-Injektionen), gefolgt von 30 mg alle 4 Wochen (Q4W).
Die empfohlene Dosis für Patienten mit einem Gewicht von ≥ 90 kg ist eine Anfangsdosis von 60 mg (zwei 30-mg-Injektionen), gefolgt von 60 mg alle 4 Wochen (Q4W).
Bei Patienten, die nach 16 Wochen Behandlung der Prurigo nodularis kein adäquates Pruritus-Ansprechen gezeigt haben, sollte ein Abbruch der Behandlung in Betracht gezogen werden.
Ausgelassene Dosis
Wenn eine Dosis ausgelassen wurde, sollte sie so bald wie möglich nachgeholt werden. Danach sollte die Dosisgabe zum geplanten Zeitpunkt wieder aufgenommen werden.
Besondere Patientengruppen
Ältere Patienten (≥ 65 Jahre)
Für ältere Patienten wird keine Dosisanpassung empfohlen (siehe Abschnitt 5.2).
Leber- und Nierenfunktionsstörung
Bei Patienten mit Leber- oder Nierenfunktionsstörung ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
Atopische Dermatitis
Die Sicherheit und Wirksamkeit von Nemolizumab bei Kindern unter 12 Jahren mit einem Körpergewicht von < 30 kg sind bisher noch nicht erwiesen. Es liegen keine Daten vor.
Prurigo nodularis
Die Sicherheit und Wirksamkeit von Nemolizumab bei Kindern unter 18 Jahren sind nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Subkutane Anwendung.
Die subkutane Injektion sollte oben vorne in den Oberschenkel oder in den Bauch verabreicht werden, wobei ein Bereich von 5 cm um den Bauchnabel auszusparen ist. Die Injektion in den Oberarm darf nur von einer Pflegeperson oder einer medizinischen Fachkraft durchgeführt werden.
Für nachfolgende Dosen wird empfohlen, die Injektionsstelle bei jeder Dosis zu wechseln. Nemolizumab darf nicht in Hautbereiche injiziert werden, die schmerzempfindlich, entzündet, geschwollen, geschädigt sind oder blaue Flecken, Narben oder offene Wunden aufweisen.
Nemolizumab ist für die Anwendung unter Anleitung eines Arztes bestimmt. Ein Patient kann sich Nemolizumab selbst injizieren oder die Pflegeperson des Patienten kann Nemolizumab verabreichen, wenn der behandelnde Arzt dies für angemessen hält. Vor der ersten Injektion sollen die Patienten und/oder Pflegepersonen eine entsprechende Schulung zur Vorbereitung und Verabreichung von Nemolizumab entsprechend der Gebrauchsanleitung am Ende der Packungsbeilage erhalten.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Überempfindlichkeit
Es wurden Fälle von Überempfindlichkeit vom Typ 1, einschließlich Angioödem, berichtet. Wenn eine systemische Überempfindlichkeitsreaktion (sofort oder verzögert) auftritt, muss die Verabreichung von Nemolizumab sofort abgebrochen und eine geeignete Therapie eingeleitet werden (siehe Abschnitt 4.8).
Verschlechterung des Asthmas (einschließlich Abnahme des exspiratorischen Spitzenflusses)
Bei der Population der Studienteilnehmer mit PN und vorbestehendem Asthma wurde nach Beginn der Behandlung mit Nemolizumab über eine leichte bis mittelschwere Verschlechterung des Asthmas (Worsening of Asthma, WOA) berichtet. Dies wurde häufiger bei Patienten mit einem Körpergewicht von > 90 kg beobachtet, die alle 4 Wochen 60 mg Nemolizumab erhielten, im Vergleich zu Patienten mit einem Körpergewicht von < 90 kg, die alle 4 Wochen 30 mg Nemolizumab erhielten (siehe Abschnitt 4.8).
Patienten mit einer Asthma-Exazerbation, die in den vorangegangenen 12 Monaten einen Krankenhausaufenthalt erforderlich machte, Patienten mit unkontrolliertem Asthma in den vorangegangenen 3 Monaten und Patienten mit einer aktuellen medizinischen Vorgeschichte von COPD und/oder chronischer Bronchitis wurden von den klinischen Studien ausgeschlossen. Es liegen keine Informationen über die Wirksamkeit oder Sicherheit von Nemolizumab bei diesen Patienten vor.
Impfungen
Es wird empfohlen, dass Patienten alle altersgemäßen Impfungen in Übereinstimmung mit den aktuellen Impfrichtlinien vor Beginn der Behandlung abschließen. Die gleichzeitige Anwendung von Lebendimpfstoffen bei Patienten, die mit Nemolizumab behandelt werden, soll vermieden werden. Es ist nicht bekannt, ob die Verabreichung von Lebendimpfstoffen während der Behandlung die Sicherheit oder Wirksamkeit dieser Impfstoffe beeinflusst. Es liegen keine Daten über die Reaktion auf Totimpfstoffe vor.
Lebendimpfstoffe
Die Sicherheit und Wirksamkeit der gleichzeitigen Anwendung von Nemolizumab mit attenuierten Lebendimpfstoffen wurde nicht untersucht. Lebendimpfstoffe sollen nicht gleichzeitig mit Nemolizumab verabreicht werden (siehe Abschnitt 4.4).
Totimpfstoffe
Die Sicherheit und Wirksamkeit der gleichzeitigen Anwendung von Nemolizumab mit Totimpfstoffen wurde nicht untersucht (siehe Abschnitt 4.4)
Wechselwirkungen mit Cytochrom P450
Die Wirkungen von Nemolizumab auf die Pharmakokinetik von Midazolam (CYP3A4/5-Substrat), Warfarin (CYP2C9-Substrat), Omeprazol (CYP2C19-Substrat), Metoprolol (CYP2D6-Substrat) und Koffein (CYP1A2-Substrat) wurden in einer Studie bei Patienten mit mittelschwerer bis schwerer AD untersucht. Es wurden keine klinisch signifikanten Veränderungen der Exposition gegenüber CYP450-Substraten im Vergleich zu vor der Nemolizumab-Behandlung beobachtet. Es ist keine Dosisanpassung erforderlich.
Schwangerschaft
Bisher liegen nur sehr begrenzte Erfahrungen mit der Anwendung von Nemolizumab bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe Abschnitt 5.3). Aus Vorsichtsgründen soll eine Anwendung von Nemolizumab während der Schwangerschaft vermieden werden.
Stillzeit
Es gibt keine Informationen darüber, ob Nemolizumab in die Muttermilch übergeht. Beim Menschen kommt es in den ersten Tagen nach der Geburt zur Ausscheidung von IgG-Antikörpern in die Muttermilch, die bald nach der Geburt auf niedrige Konzentrationen absinken. Infolgedessen kann es in den ersten Tagen zu einer Übertragung von IgG-Antikörpern auf das Neugeborene über die Muttermilch kommen. In diesem kurzen Zeitraum kann ein Risiko für das gestillte Kind nicht ausgeschlossen werden. Danach könnte Nemolizumab während der Stillzeit angewendet werden, wenn dies aus klinischer Sicht notwendig ist.
Fertilität
Tierexperimentelle Studien haben keine Beeinträchtigung der Fertilität gezeigt (siehe Abschnitt 5.3).
Nemluvio hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen bei atopischer Dermatitis und Prurigo nodularis sind Typ-I-Überempfindlichkeitsreaktionen (1,1 %; umfasst Urtikaria 1,0 % und Angioödem 0,1 %) und Reaktionen an der Injektionsstelle (1,2 %) (siehe Abschnitt 4.4). Weitere Nebenwirkungen wie Kopfschmerzen (7,0 %), atopische Dermatitis (4,6 %), Ekzem (3,8 %) und nummuläres Ekzem (3,5 %) wurden bei Prurigo nodularis berichtet.
Tabellarische Auflistung der Nebenwirkungen
In Tabelle 1 sind alle in klinischen Studien beobachteten Nebenwirkungen nach Systemorganklasse und Häufigkeit unter Verwendung der folgenden Kategorien aufgeführt: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1 000, < 1/100); selten (≥ 1/10 000, < 1/1 000); sehr selten (< 1/10 000). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad geordnet.
Tabelle 1: Auflistung der Nebenwirkungen
MedDRA-Systemorganklasse | Häufigkeit | Nebenwirkungen |
Infektionen und parasitäre Erkrankungen | Häufig | Oberflächliche Pilzinfektionen*# |
Erkrankungen des Blutes und des Lymphsystems | Gelegentlich | Eosinophilie† |
Erkrankungen des Immunsystems | Häufig | Typ-I-Allergie (einschl. Urtikaria† und Angioödem*) |
Erkrankungen des Nervensystems | Häufig | Kopfschmerzen* (einschl. Spannungskopfschmerzen) |
Erkrankungen der Atemwege, des Brustraums und Mediastinums | Häufig | Verschlechterung des Asthmas* (einschließlich Asthma, Giemen, exspiratorischer Spitzenfluss erniedrigt) |
Erkrankungen der Haut und des Unterhautgewebes | Häufig | Atopische Dermatitis*, |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Häufig | Reaktionen an der Injektionsstelle (einschl. Erythem, Pruritus, Hämatom†, Schmerz†, Reizung†, blaue Flecken* und Ödem an der Injektionsstelle†) |
†Aufgetreten in Studien zu atopischer Dermatitis
*Aufgetreten in Studien zu Prurigo nodularis
#Zu den oberflächlichen Pilzinfektionen gehören: Tinea corporis, Tinea pedis, Onychomykose, Pilzinfektion, Tinea versicolor, Tinea cruris, Hautpilzinfektion und Fußpilzinfektion
Beschreibung ausgewählter Nebenwirkungen
Überempfindlichkeit
Überempfindlichkeitsreaktionen vom Typ 1 (Ig-E-vermittelte Reaktionen), einschließlich leichter Urtikaria und leichter (periokulärer) Angioödeme im Gesicht, wurden in den klinischen Studien häufig bei Patienten beobachtet, die mit Nemolizumab behandelt wurden. Diese Reaktionen führten nicht zum Abbruch der Behandlung (siehe Abschnitt 4.4).
Kopfschmerzen
Bei Patienten mit Prurigo nodularis wurden Kopfschmerzen bei Patienten, die mit Nemolizumab behandelt wurden (7,0 %), häufiger berichtet als bei Patienten, die mit Placebo behandelt wurden. In beiden Gruppen wurden Kopfschmerzen bei Frauen häufiger beobachtet. In der Nemolizumab‑Gruppe waren Kopfschmerzen meist leicht oder mittelschwer und führten nicht zum Abbruch der Behandlung.
Verschlechterung des Asthmas
Bei PN-Patienten mit vorbestehendem Asthma (n = 51) kam es bei 8 (15,7 %) Patienten nach Beginn der Behandlung mit Nemolizumab zu einer Verschlechterung des Asthmas (Worsening of Asthma, WOA), von denen 5 ein Körpergewicht von > 90 kg hatten und alle 4 Wochen 60 mg Nemolizumab erhielten. In der Population der PN-Patienten mit vorbestehendem Asthma war WOA bei Patienten mit einem Körpergewicht von > 90 kg, die alle 4 Wochen 60 mg Nemolizumab erhielten, dreimal häufiger als bei Patienten mit einem Körpergewicht von < 90 kg, die alle 4 Wochen 30 mg Nemolizumab erhielten.
Die meisten WOA-Ereignisse traten innerhalb der ersten zwei Monate nach Beginn der Behandlung auf und alle wurden als leicht oder mittelschwer gemeldet. Bei den meisten Patienten trat während der Behandlung ein einzelnes WOA-Ereignis auf, das mit Standard-Asthmamedikamenten (Inhalatoren) ohne systemische Steroide behandelt werden konnte. Keines der Ereignisse führte zu einem dauerhaften Abbruch der Behandlung. Die Inzidenz von WOA stieg in der offenen Langzeitverlängerungsstudie bei PN mit längerfristiger Exposition gegenüber Nemolizumab (bis Woche 52) nicht an.
Ekzematöse Reaktionen
Bei Patienten mit Prurigo nodularis wurden ekzematöse Reaktionen wie atopische Dermatitis, nummuläres Ekzem oder Ekzem bei mit Nemolizumab behandelten Patienten häufiger berichtet als bei Patienten, die mit Placebo behandelt wurden: Atopische Dermatitis (4,6 %), Ekzem (3,8 %) und nummuläres Ekzem (3,5 %). Diese ekzematösen Reaktionen waren leicht oder mittelschwer. Atopische Dermatitis führte bei 2 (0,5 %) Patienten zum Absetzen von Nemolizumab. Bei Patienten im Alter von > 65 Jahren war die Rate ekzematöser Reaktionen höher.
Eosinophilie
Der Anteil der Patienten mit klinisch signifikant erhöhten Eosinophilen (> 700 Zellen/mcl) betrug 10,2 % in der AD-Population (in der Anfangsphase) und 5,5 % in der PN-Population. Eine schwere Eosinophilie (> 5000 Zellen/mcl) wurde bei AD-Patienten, die mit Nemolizumab behandelt wurden, in der ersten Behandlungsphase nicht beobachtet. Unerwünschte Reaktionen in Form von Eosinophilie wurden bei 0,2 % der AD-Patienten, die während der ersten Behandlungsphase bis Woche 16 mit Nemolizumab behandelt wurden, gemeldet. Alle Ereignisse bei den AD-Studienteilnehmern waren von leichter Intensität und nicht mit klinischen Symptomen verbunden. Kein therapieassoziiertes unerwünschtes Ereignis von Eosinophilie führte zum Abbruch der Behandlung. Abgesehen von einem Fall von eosinophiler Kolitis bei einem AD-Studienteilnehmer mit anderen atopischen Komorbiditäten gab es keine weiteren Berichte über eosinophile Störungen.
Kinder und Jugendliche
Atopische Dermatitis
Jugendliche (12 bis 17 Jahre)
Die Sicherheit von Nemolizumab wurde bei 176 pädiatrischen Patienten im Alter von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis untersucht, die in die Studien ARCADIA 1 und ARCADIA 2 aufgenommen wurden. Das Sicherheitsprofil von Nemolizumab bei diesen Patienten war bis Woche 16 vergleichbar dem Sicherheitsprofil, das bei Erwachsenen mit atopischer Dermatitis beobachtet wurde.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51-59, 63225 Langen, Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
Es gibt keine spezifische Behandlung für eine Nemolizumab-Überdosierung. Im Falle einer Überdosierung soll der Patient auf Symptome von Nebenwirkungen überwacht werden, und es soll umgehend eine geeignete symptomatische Behandlung eingeleitet werden.
Pharmakotherapeutische Gruppe: Andere Dermatika, Mittel zur Behandlung der Dermatitis, exkl. Corticosteroide, ATC-Code: D11AH12
Wirkmechanismus
Nemolizumab ist ein humanisierter monoklonaler IgG2-Antikörper, der die Interleukin-31-(IL-31-)-Signaltransduktion hemmt, indem er selektiv an den Interleukin-31-Rezeptor alpha (IL-31 RA) bindet. IL-31 ist ein natürlich vorkommendes Zytokin, das an Pruritus, Entzündung, epidermaler Dysregulation und Fibrose beteiligt ist. Nemolizumab hemmt IL-31-induzierte Reaktionen, einschließlich der Freisetzung von proinflammatorischen Zytokinen und Chemokinen.
In klinischen Studien zu atopischer Dermatitis wurde festgestellt, dass Nemolizumab die Genexpression moduliert, die mit der Pathophysiologie der atopischen Dermatitis zusammenhängt, mit primärer Wirkung auf die Prozesse des Immunsystems, indem es das inflammatorische und proliferative Profil spezifischer Immunzellen (T-Zellen und Monozyten/Makrophagen) reduziert, ohne zu einer Immunsuppression zu führen.
In klinischen Studien zu Prurigo nodularis wurde festgestellt, dass Nemolizumab molekulare Prozesse moduliert, die mit der Pathophysiologie von Prurigo nodularis zusammenhängen, mit Auswirkungen auf Pruritus, Entzündung, epidermale Differenzierung und Fibrose.
Pharmakodynamische Wirkung
Immunogenität
Anti-Wirkstoff-Antikörper (anti-drug antibodies, ADA) wurden sehr häufig nachgewiesen. Es wurden keine Anzeichen für einen ADA‑Einfluss auf die Pharmakokinetik, Wirksamkeit oder Sicherheit beobachtet.
Klinische Wirksamkeit und Sicherheit bei atopischer Dermatitis
Erwachsene und Jugendliche mit atopischer Dermatitis
Die Wirksamkeit und Sicherheit von Nemolizumab mit begleitender topischer Hintergrundtherapie wurde in zwei randomisierten, doppelblinden, placebokontrollierten Zulassungsstudien (ARCADIA 1 und ARCADIA 2) untersucht, in die insgesamt 1 728 Studienteilnehmer ab 12 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis aufgenommen wurden, die durch topische Behandlungen nicht ausreichend kontrolliert werden konnte. Der Schweregrad der Erkrankung wurde durch einen IGA-(Investigator's Global Assessment)Score von 3 (mittelschwer) und 4 (schwer) in der Gesamtbeurteilung der atopischen Dermatitis, einen EASI-(Eczema Area and Severity Index)Score von ≥ 16, eine minimale betroffene Körperoberfläche von ≥ 10 % und einen PP-NRS-(Peak Pruritus Numeric Rating Scale)Score von ≥ 4 definiert.
Die Patienten in den Studien erhielten entweder eine erste subkutane Injektion von Nemolizumab 60 mg gefolgt von 30 mg Injektionen alle 4 Wochen (Q4W) oder entsprechendes Placebo. Begleitende TCS mit niedriger und/oder mittlerer Wirkstärke und/oder TCI wurden sowohl in den Nemolizumab, als auch in den Placebo-Gruppen für mindestens 14 Tage vor Baseline verabreicht und während der Studie fortgesetzt. Je nach Krankheitsaktivität konnten diese Begleittherapien nach Ermessen des Prüfarztes ausgeschlichen und/oder abgesetzt werden.
Nach 16 Wochen fuhren die Patienten, die entweder EASI-75 oder IGA erreicht hatten, für weitere 32 Wochen mit dem Erhaltungszeitraum der Studie fort, um die Aufrechterhaltung des in Woche 16 erreichten Ansprechens zu untersuchen. Die Nemolizumab-Responder wurden erneut randomisiert und erhielten entweder 30 mg Nemolizumab alle 4 Wochen, 30 mg Nemolizumab alle 8 Wochen oder Placebo alle 4 Wochen (alle Gruppen setzten die Hintergrundtherapie mit TCS/TCI fort). Patienten, die im anfänglichen Behandlungszeitraum auf Placebo randomisiert wurden und in Woche 16 das gleiche klinische Ansprechen erreichten, erhielten weiterhin Placebo alle 4 Wochen. Non-Responder in Woche 16, Patienten, deren klinisches Ansprechen während des Erhaltungszeitraums verloren ging, und Patienten, die den Erhaltungszeitraum abschlossen, hatten die Möglichkeit, in die offene Studie (ARCADIA LTE) aufgenommen zu werden und bis zu 200 Wochen lang alle 4 Wochen eine Behandlung mit Nemolizumab 30 mg zu erhalten.
Endpunkte
Sowohl ARCADIA 1 als auch ARCADIA 2 untersuchte die folgenden primären Endpunkte:
Anteil der Patienten mit IGA-Erfolg (definiert als IGA 0 [erscheinungsfrei] oder 1 [beinahe erscheinungsfrei] und Reduktion um ≥ 2 Punkte gegenüber Baseline) in Woche 16
Anteil der Patienten mit EASI-75 (≥ 75 % Verbesserung des EASI gegenüber Baseline) in Woche 16
Sekundäre Hauptendpunkte waren PP-NRS-Verbesserung ≥ 4 gegenüber Baseline in den Wochen 1, 2, 4 und 16, PP-NRS < 2 in Woche 4 und Woche 16, Verbesserung auf der SD-NRS (Sleep Disturbance Numeric Rating Scale) ≥ 4 gegenüber Baseline in Woche 16, Patienten mit einer Verbesserung sowohl des EASI-75 als auch des PP-NRS ≥ 4 gegenüber Baseline in Woche 16 und Patienten mit sowohl IGA-Erfolg als auch Verbesserung des PP-NRS ≥ 4 gegenüber Baseline in Woche 16.
Baseline-Merkmale
In diesen Studien waren zu Studienbeginn 51,0 % der Patienten männlich, 79,9 % waren kaukasisch und das mittlere Gewicht betrug 75,0 kg. Das Durchschnittsalter lag bei 34,1 Jahren, 15,4 % der Patienten waren Jugendliche (12–17 Jahre) und 5,3 % waren 65 Jahre alt oder älter. 70 % der Patienten hatten einen Baseline-IGA-Score von 3 (mittelschwere AD) und 30 % der Patienten hatten einen Baseline-IGA-Score von 4 (schwere AD). Der mittlere (SD) EASI-Ausgangswert betrug 27,5 (10,5), der wöchentliche durchschnittliche (SD) PP-NRS-Ausgangswert betrug 7,1 (1,5) (schwerer Juckreiz) und der wöchentliche durchschnittliche (SD) SD-NRS-Ausgangswert betrug 5,8 (2,2). Insgesamt erhielten 63,3 % der Patienten andere vorherige systemische Behandlungen für atopische Dermatitis.
Klinisches Ansprechen
ARCADIA 1 und ARCADIA 2 – Erwachsene und Jugendliche – Induktionsphase, Woche 0 bis Woche 16
Nemolizumab war Placebo hinsichtlich der hautbezogenen co-primären Endpunkte IGA-Erfolg und EASI-75 über 16 Wochen statistisch signifikant überlegen (Tabelle 2). Die Ergebnisse für beide co-primären Endpunkte waren in der Population mit schwerem Pruritus konsistent (Baseline-PP-NRS ≥ 7).
Tabelle 2 – Wirksamkeitsergebnisse für Nemolizumab (30 mg Q4W) mit begleitendem TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16
ARCADIA 1 | ARCADIA 2 | |||
Nemolizumab + TCS/TCI | Placebo + | Nemolizumab + TCS/TCI | Placebo + | |
Anzahl der randomisierten und behandelten Patienten (Baseline PP-NRS ≥ 4) | 620 | 321 | 522 | 265 |
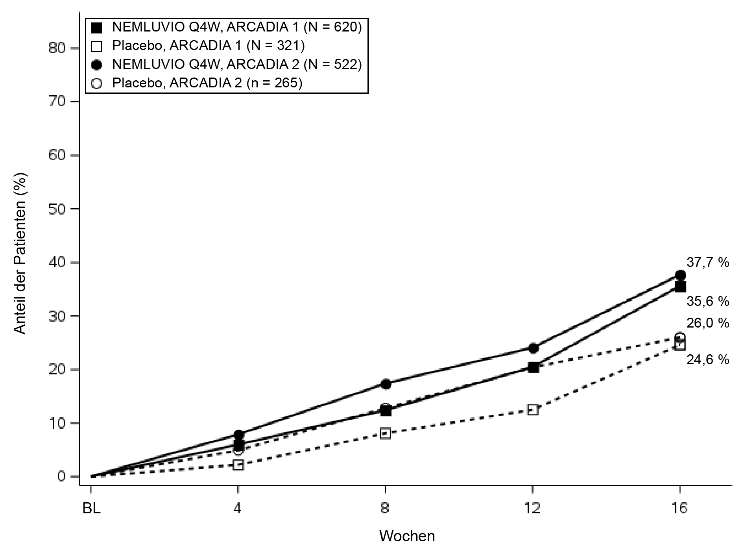
% der Patienten mit IGA 0 oder 1a | 35,6# | 24,6 | 37,7# | 26,0 |
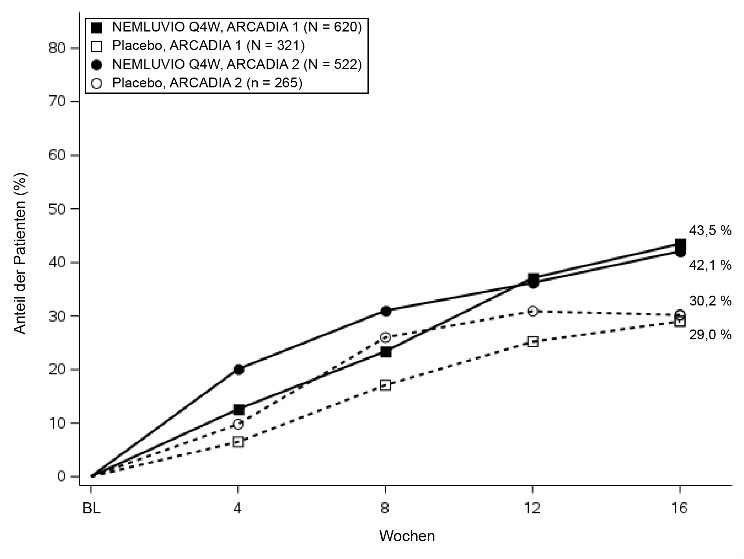
% der Patienten mit EASI-75a | 43,5* | 29,0 | 42,1# | 30,2 |
a Patienten, die eine Rescue-Therapie erhielten oder für die Daten fehlten, galten als Non-Responder
*p-Wert < 0,0001, #p-Wert < 0,001
Der p-Wert nach Strata basiert auf dem CMH-Test, stratifiziert nach PP-NRS und IGA-Score bei Baseline
Abbildung 1 – Anteil der Patienten mit IGA-Erfolg und EASI-75 von Baseline bis Woche 16 in ARCADIA 1 und ARCADIA 2
Abbildung 1a. IGA-Erfolg |
Abbildung 1b. EASI-75 |
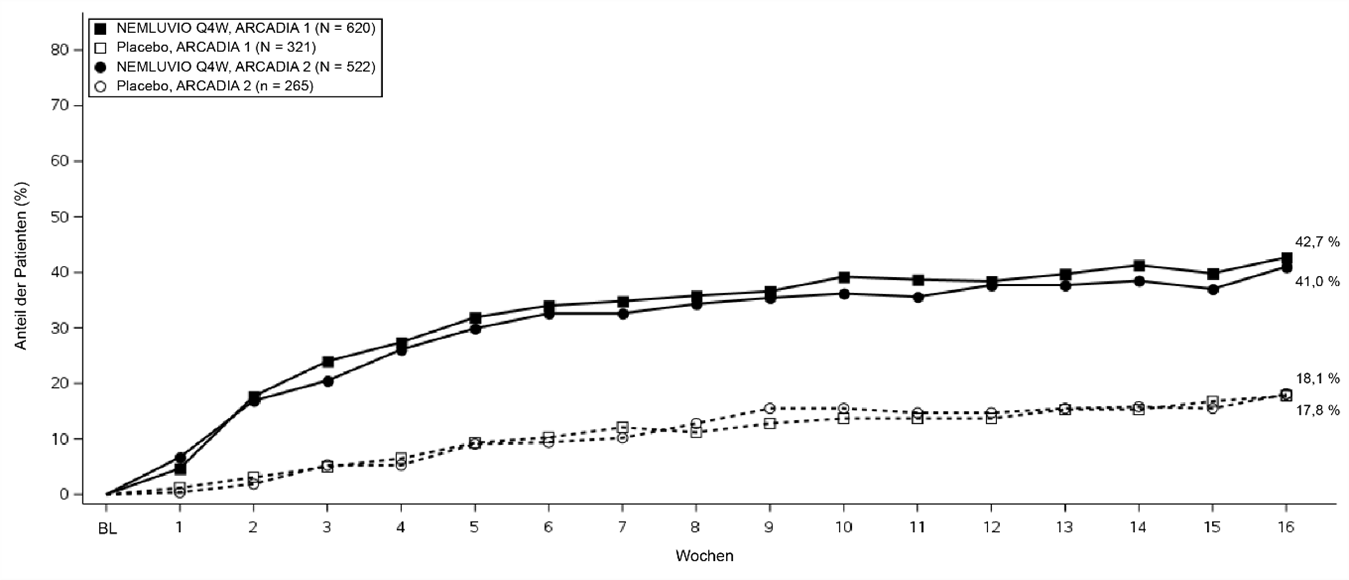
Eine signifikante Besserung des Pruritus bei Patienten, die in ARCADIA 1 und ARCADIA 2 mit Nemolizumab behandelt wurden, im Vergleich zu Placebo, basierend auf PP-NRS-Verbesserungen ≥ 4 und prozentualer Veränderung auf der PP-NRS gegenüber Baseline wurde ab Woche 1 beobachtet und blieb bis Woche 16 erhalten (Tabelle 3 und Abbildung 2). Die Ergebnisse waren in der Population mit schwerem Pruritus konsistent (Baseline-PP-NRS ≥ 7).
Tabelle 3 – Wirksamkeitsergebnisse zu Juckreiz bei Nemolizumab mit begleitendem TCS/TCI in ARCADIA 1 und ARCADIA 2 bis Woche 16
ARCADIA 1 | ARCADIA 2 | |||
Nemolizumab + TCS/TCI | Placebo + TCS/TCI | Nemolizumab + TCS/TCI | Placebo + TCS/TCI | |
Anzahl der randomisierten und behandelten Patienten (Baseline PP-NRS ≥ 4)a | 620 | 321 | 522 | 265 |
% der Patienten mit PP-NRS-Verbesserung ≥ 4a | ||||
In Woche 1 | 4,7§ | 1,2 | 6,7* | 0,4 |
In Woche 2 | 17,7* | 3,1 | 16,9* | 1,9 |
In Woche 4 | 27,4* | 6,5 | 26,1* | 5,3 |
In Woche 16 | 42,7* | 17,8 | 41,0* | 18,1 |
% der Patienten mit PP-NRS < 2a | ||||
In Woche 4 | 16,0* | 3,7 | 15,9* | 2,6 |
In Woche 16 | 30,6* | 11,2 | 28,4* | 11,3 |
Mittlere Veränderung gegenüber dem Ausgangswert (%) | ||||
In Woche 16 | -56,1* | -30,6 | -55,6* | -30,3 |
a Patienten, die eine Rescue-Therapie erhielten oder für die Daten fehlten, galten als Non-Responder
*p-Wert < 0,0001, §p-Wert < 0,05
Der p-Wert nach Strata basiert auf dem CMH-Test, stratifiziert nach PP-NRS und IGA-Score bei Baseline
Abbildung 2 –Anteil der Patienten mit PP-NRS-Verbesserung von ≥ 4 von Baseline bis Woche 16 in ARCADIA 1 und ARCADIA 2
Bei Patienten mit einem Körpergewicht von ≥ 90 kg gab es in einer Post-hoc-Analyse in jeder der Zulassungsstudien in Woche 16 keinen Unterschied im antiinflammatorischen Ansprechen (IGA 0 oder 1 und EASI-75) zwischen den Nemolizumab- und den Placebo-Armen, obwohl die Wirkung bei der Verringerung des Juckreizes (PP NRS) beobachtet wurde.
Die numerische Rating-Skala für Schlafstörungen (SD-NRS, Sleep Disturbance Numeric Rating Scale) ist eine Skala, anhand derer die Patienten täglich den Grad ihres Schlafverlusts im Zusammenhang mit atopischer Dermatitis berichten. Eine signifikante Besserung der Schlafstörungen im Vergleich zu Placebo wurde in Woche 16 beobachtet (Tabelle 4). Die Ergebnisse waren in der Population mit schwerem Pruritus konsistent (Baseline-PP-NRS ≥ 7).
Tabelle 4 – Wirksamkeit bei Schlafstörungen für Nemolizumab mit begleitendem TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16
ARCADIA 1 | ARCADIA 2 | |||
Nemolizumab + TCS/TCI | Placebo + TCS/TCI | Nemolizumab + TCS/TCI | Placebo + TCS/TCI | |
Anzahl der randomisierten und behandelten Patienten (Baseline PP-NRS ≥ 4)a | 620 | 321 | 522 | 265 |
% der Patienten mit SD-NRS-Verbesserung ≥ 4a | 37,9* -64,6 | 19,9 -38,1 | 33,5* -59,7 | 16,2 -35,4 |
a Patienten, die eine Rescue-Therapie erhielten oder für die Daten fehlten, galten als Non-Responder
*p-Wert < 0,0001
Der p-Wert nach Strata basiert auf dem CMH-Test, stratifiziert nach PP-NRS und IGA-Score bei Baseline
Jugendliche mit atopischer Dermatitis (12 bis 17 Jahre)
Die Wirksamkeitsergebnisse aus den Studien ARCADIA 1 und ARCADIA 2 in Woche 16 für pädiatrische Patienten im Alter von 12 bis 17 Jahren sind in Tabelle 5 dargestellt. Die Ergebnisse in der pädiatrischen Patientenpopulation stimmten im Allgemeinen mit den Ergebnissen in der erwachsenen Patientenpopulation überein. Die Ergebnisse der co-primären und sekundären Hauptendpunkte waren in der Population mit schwerem Pruritus konsistent (Baseline-PP-NRS ≥ 7).
Tabelle 5 – Wirksamkeitsergebnisse für Nemolizumab (30 mg Q4W) mit begleitendem TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 16 bei pädiatrischen Patienten im Alter von 12 bis 17 Jahren
ARCADIA 1 UND ARCADIA 2 | ||
Nemolizumab + TCS/TCI | Nemolizumab + TCS/TCI | |
Anzahl der randomisierten und behandelten Patienten (Baseline PP-NRS ≥ 4) | 179 | 90 |
% der Patienten mit IGA 0 oder 1 a | 48,9* | 34,4 |
% der Patienten mit EASI-75 a | 53,4§ | 43,3 |
% der Patienten mit PP-NRS-Verbesserung ≥ 4 a | 40,9# | 17,8 |
% der Patienten mit PP-NRS < 2 a | 30,1≠ | 6,7 |
% der Patienten mit SD-NRS-Verbesserung ≥ 4 a | 31,8∞ | 20,0 |
a Patienten, die eine Rescue-Therapie erhielten oder für die Daten fehlten, galten als Non-Responder
≠p-Wert < 0,0001, #p-Wert < 0,001, *p-Wert < 0,05, ∞p-Wert = 0,0591, §p-Wert = 0,1824
Der p-Wert nach Strata basiert auf dem CMH-Test, stratifiziert nach PP-NRS und IGA-Score bei Baseline
ARCADIA 1 und ARCADIA 2 – Erwachsene und Jugendliche – Erhaltungszeitraum, Woche 16 bis Woche 48
Das klinische Ansprechen bei Nemolizumab-Respondern (IGA 0/1 oder EASI-75 in Woche 16) wurde in den Studien ARCADIA 1 und ARCADIA 2 zwischen Woche 16 und Woche 48 beurteilt. Für den Zeitraum der Erhaltungstherapie wurden 507 Nemolizumab-Responder erneut randomisiert und erhielten entweder Nemolizumab 30 mg Q4W, Nemolizumab 30 mg Q8W oder Placebo Q4W (Ausschleichen von Nemolizumab) mit begleitendem TCS/TCI. Die gepoolten Wirksamkeitsergebnisse mit deskriptiver Analyse nur für diesen Zeitraum in den Zulassungsstudien (ARCADIA 1 und ARCADIA 2) mit Nemolizumab in Woche 48 sind in Tabelle 6 dargestellt.
Tabelle 6 –Gepoolte Wirksamkeitsergebnisse für den Erhaltungszeitraum für Nemolizumab mit begleitendem TCS/TCI in ARCADIA 1 und ARCADIA 2 in Woche 48
Nemolizumab + TCS/TCI | Nemolizumab + TCS/TCI | Placebo + TCS/TCI | |
% der Patienten mit IGA 0 oder 1a | |||
Woche 16 (Beginn Erhaltungstherapie) | 84,0 | 84,0 | 77,5 |
Woche 48 | 61,5 | 60,4 | 49,7 |
% der Patienten mit EASI-75a (95-%-KI) | |||
Woche 16 (Beginn Erhaltungstherapie) | 96,4 | 96,4 | 92,9 |
Woche 48 | 76,3 | 75,7 | 63,9 |
a Patienten, die eine Rescue-Therapie erhielten oder für die Daten fehlten, galten als Non-Responder
Klinische Wirksamkeit und Sicherheit bei Erwachsenen mit Prurigo nodularis
Die Wirksamkeit und Sicherheit von Nemolizumab als Monotherapie wurde in zwei randomisierten, doppelblinden, placebokontrollierten Zulassungsstudien (OLYMPIA 1 und OLYMPIA 2) untersucht, in die insgesamt 560 Patienten ab 18 Jahren mit mittelschwerer bis schwerer Prurigo nodularis aufgenommen wurden. Der Schweregrad der Erkrankung wurde anhand der IGA-(Investigator's Global Assessment)Skala zur Gesamtbeurteilung der Prurigo-nodularis-Läsionen auf einer Schweregradskala von 0 bis 4 definiert. Die in diese beiden Studien aufgenommenen Patienten hatten einen IGA-Score von ≥ 3, schweren Pruritus, definiert als wöchentlicher Durchschnitt des PP-NRS-(Peak Pruritus Numeric Rating Scale)Scores von ≥ 7 auf einer Skala von 0 bis 10 und mindestens 20 noduläre Läsionen. OLYMPIA 1 und OLYMPIA 2 untersuchten die Wirkung der Nemolizumab-Monotherapie auf die Prurigo-nodularis-Symptome und zielten auf eine Besserung der Hautläsionen und des Pruritus über 16 Wochen ab. OLYMPIA 1 hatte einen 24-wöchigen Behandlungszeitraum und OLYMPIA 2 einen 16-wöchigen Behandlungszeitraum.
In der Nemolizumab-Behandlungsgruppe erhielten Patienten mit einem Gewicht von weniger als 90 kg subkutane Injektionen von Nemolizumab 60 mg (2 Injektionen zu 30 mg) in Woche 0, gefolgt von 30-mg-Injektionen alle 4 Wochen, und Patienten mit einem Gewicht von 90 kg oder mehr erhielten subkutane Injektionen von Nemolizumab 60 mg (2 Injektionen zu 30 mg) in Woche 0 und alle 4 Wochen.
Endpunkte
Sowohl OLYMPIA 1 als auch OLYMPIA 2 bewerteten dieselben zwei primären Endpunkte:
Anteil der Patienten mit einer Verbesserung von ≥ 4 gegenüber Baseline in der PP-NRS (Peak Pruritus Numeric Rating Scale) in Woche 16
Anteil der Patienten mit IGA-Erfolg (definiert als IGA von 0 [erscheinungsfrei] oder 1 [beinahe erscheinungsfrei] und einer Verbesserung um ≥ 2 Punkte gegenüber Baseline) in Woche 16
Sekundäre Hauptendpunkte waren eine PP-NRS-Verbesserung ≥ 4 von Baseline bis Woche 4, eine PP-NRS < 2 in Woche 4 und Woche 16, eine Verbesserung in der SD-NRS (Sleep Disturbance Numeric Rating Scale) ≥ 4 gegenüber Baseline in Woche 4 und 16.
Baseline-Merkmale
In diesen Studien waren vor Behandlungsbeginn 59,6 % der Patienten weiblich, 81,4 % waren kaukasisch, das mittlere Gewicht betrug 82,6 kg, das mittlere Alter lag bei 55,2 Jahren und 25,4 % der Patienten waren älter als 65 Jahre. Der wöchentliche durchschnittliche PP-NRS-Score bei Baseline lag im Mittel (SD) bei 8,4 (0,9). Achtundfünfzig (58) Prozent der Patienten hatten einen Baseline-IGA-Score von 3 (mittelschwere PN) und 42 % der Patienten hatten einen Baseline-IGA von 4 (schwere PN).
Klinisches Ansprechen
Pivotstudien (OLYMPIA 1 und OLYMPIA 2) – Woche 0 bis Woche 16
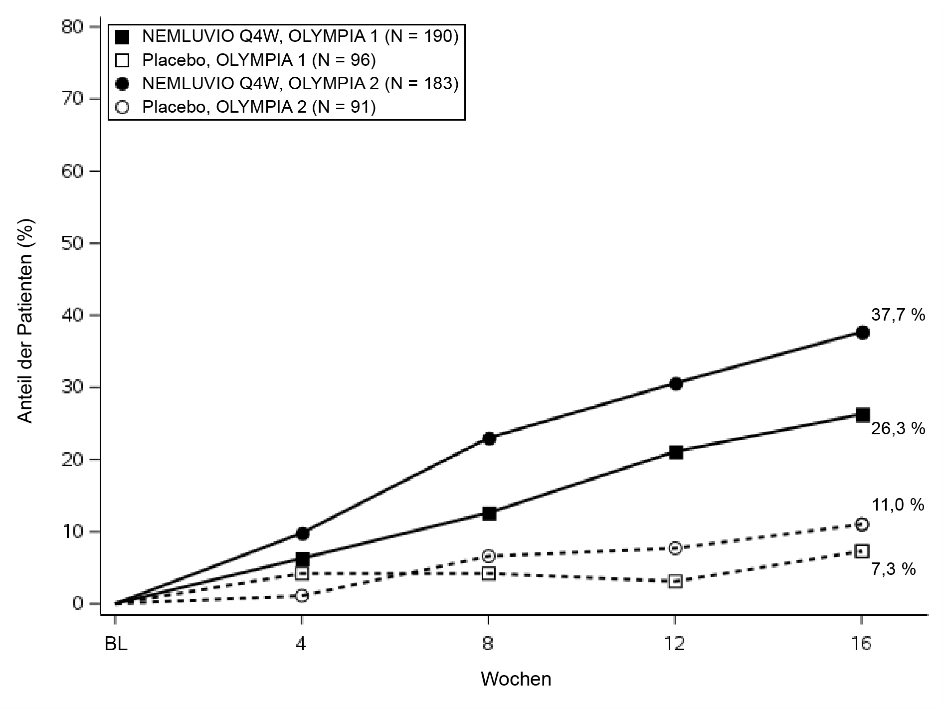
Die Ergebnisse der Zulassungsstudien zur Beurteilung der Behandlung mit Nemolizumab in OLYMPIA 1 und OLYMPIA 2 sind in Tabelle 7 dargestellt und zeigen eine signifikante Verbesserung bei mit Nemolizumab behandelten Patienten im Vergleich zu Placebo für beide primären Endpunkte (Abbildung 3 und Abbildung 4).
Tabelle 7 – Wirksamkeitsergebnisse für Nemolizumab-Monotherapie (Q4W) in OLYMPIA 1 und OLYMPIA 2
OLYMPIA 1 | OLYMPIA 2 | |||
Nemolizumab | Placebo | Nemolizumab | Placebo | |
Anzahl der randomisierten Patienten | 190 | 96 | 183 | 91 |
% der Patienten mit einer Verbesserung in der PP-NRS ≥ 4 gegenüber Baselinea | ||||
Woche 4 | 41,1* | 6,3 | 41,0* | 7,7 |
Woche 16 | 58,4* | 16,7 | 56,3* | 20,9 |
% der Patienten mit IGA 0 oder 1 in Woche 16a | 26,3# | 7,3 | 37,7* | 11 |
% der Patienten mit PP-NRS < 2 a | ||||
Woche 4 | 21,6* | 1,0 | 19,7* | 2,2 |
Woche 16 | 34,2* | 4,2 | 35,0* | 7,7 |
% der Patienten mit Verbesserung in der SD-NRS ≥ 4 gegenüber Baselinea | ||||
Woche 4 | 31,1* | 5,2 | 37,2* | 9,9 |
Woche 16 | 50,0* | 11,5 | 51,9* | 20,9 |
a Wenn ein Patient eine Rescue-Therapie erhielt, wird eine Strategie der zusammengesetzten Variablen angewendet, die zugrunde liegenden Daten bei/nach Erhalt der Rescue-Therapie werden als schlechtestmöglicher Wert angenommen und das Ansprechen wird aus dem zugrunde liegenden Datenwert abgeleitet. Patienten mit fehlenden Ergebnissen gelten als Non-Responder.
*p-Wert < 0,0001, #p-Wert = 0,0025 Strata-bereinigt anhand der randomisierten Stratifizierungsvariablen (Analysenzentrum und Körpergewicht bei Baseline (< 90 kg, ≥ 90 kg)
Abbildung 3 – Anteil der Patienten mit PP-NRS-Verbesserung ≥ 4 von Baseline bis Woche 16
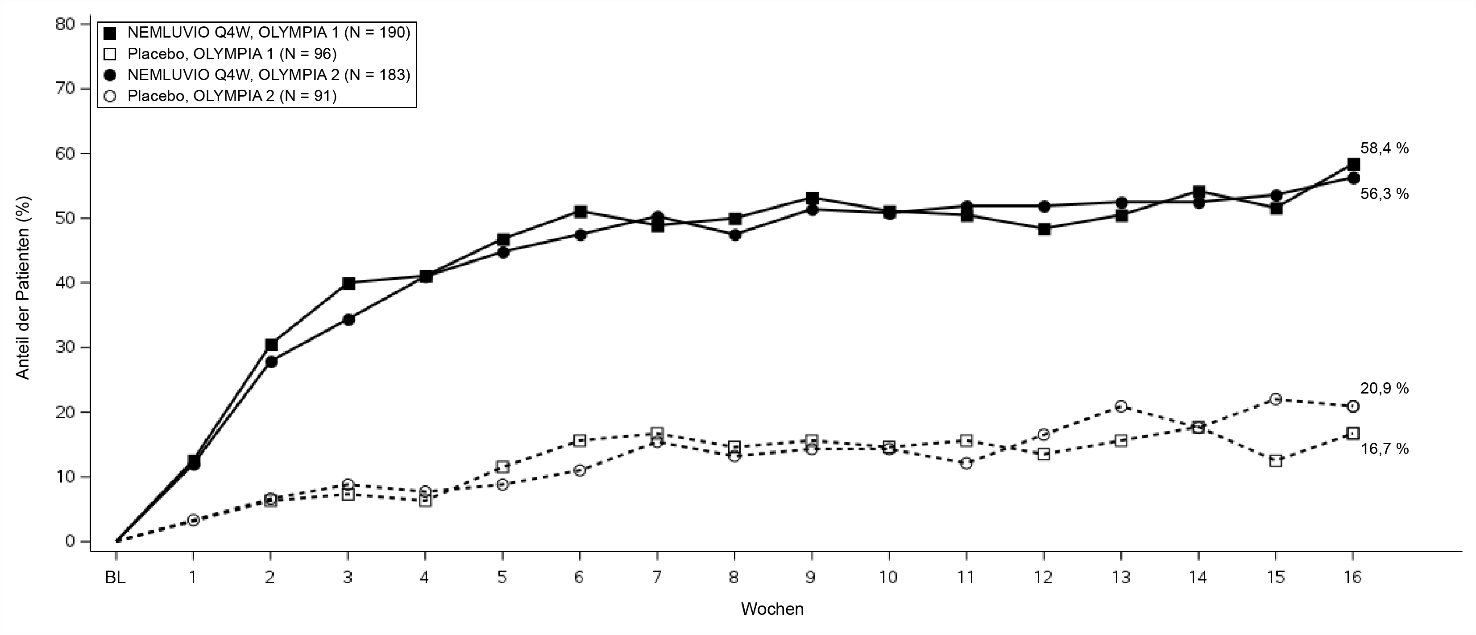
Abbildung 4 – Anteil der IGA-Responder von Baseline bis Woche 16
Resorption
Bei Patienten mit AD oder PN betrug nach einer anfänglichen subkutanen Dosis von 60 mg die geschätzte mittlere (SD) populationspharmakokinetische Spitzenkonzentration (Cmax) 6,7 (2,20) µg/ml etwa 6 Tage nach der Dosis.
Nach mehreren Dosen bei Patienten mit atopischer Dermatitis betrugen die geschätzten mittleren (SD) populationspharmakokinetischen Steady-State-Talkonzentrationen von Nemolizumab 2,63 (1,27) µg/ml für 30 mg bei Q4W-Verabreichung und 0,74 (0,44) µg/ml für 30 mg bei Q8W-Verabreichung.
Nach mehreren Dosen bei Patienten mit Prurigo nodularis betrugen die geschätzten mittleren (SD) populationspharmakokinetischen Steady-State-Talkonzentrationen von Nemolizumab 3,04 (1,23) µg/ml bei Patienten mit einem Körpergewicht von < 90 kg bei 30 mg Q4W und 3,66 (1,63) µg/ml bei Patienten mit einem Körpergewicht von ≥ 90 kg bei 60 mg Q4W.
Sowohl in der atopischen-Dermatitis- als auch in der Prurigo-nodularis-Population wurden Steady-State-Konzentrationen von Nemolizumab nach einer Aufsättigungsdosis von 60 mg in Woche 4 und ohne Aufsättigungsdosis in Woche 12 erreicht.
Eine Aufsättigungsdosis wird für Patienten mit PN mit einem Körpergewicht < 90 kg vorgeschlagen. Für Patienten mit einem Körpergewicht von ≥ 90 kg wird jedoch keine Aufsättigungsdosis vorgeschlagen, da die 60-mg-Dosis ausreichte, um vergleichbare Steady-State-Konzentrationen von Nemolizumab zu erreichen wie die 30-mg-Dosis (mit 60-mg-Aufsättigungsdosis) nach der zweiten Dosis (in Woche 8).
Verteilung
Auf der Grundlage einer populationspharmakokinetischen Analyse betrug das scheinbare Verteilungsvolumen (V/F) 7,67 l.
Biotransformation
Da es sich bei Nemolizumab um ein Protein handelt, wurden keine besonderen Studien zur Verstoffwechslung durchgeführt. Es wird erwartet, dass Nemolizumab durch katabole Stoffwechselwege zu kleinen Peptiden metabolisiert wird.
Elimination
Es wird erwartet, dass Nemolizumab auf die gleiche Weise abgebaut wird wie endogenes IgG. In der populationspharmakokinetischen Analyse wurde die terminale Halbwertszeit (SD) von Nemolizumab auf 18,9 (4,96) Tage und die scheinbare systemische Clearance (Cl/F) auf 0,26 l/Tag geschätzt.
Linearität/Nicht-Linearität
Nach einer Einzeldosis zeigte Nemolizumab eine lineare Pharmakokinetik, wobei die Expositionen zwischen 0,03 und 3 mg/kg dosisproportional anstiegen.
Nach mehreren Dosen stieg die systemische Exposition von Nemolizumab im gesamten subkutanen Dosisbereich bis zu 30 mg ungefähr dosisproportional an. Bei der subkutanen Dosis von 60 mg kam es zu einer leichten Abnahme der Bioverfügbarkeit um 9 %.
Besondere Patientengruppen
Geschlecht, Alter und ethnische Herkunft
Geschlecht, Alter (Bereich 12 bis 85 Jahre für AD und 18 bis 84 Jahre für PN) und ethnische Herkunft hatten keine klinisch relevante Wirkung auf die Pharmakokinetik von Nemolizumab.
Leberfunktionsstörung
Es wird nicht erwartet, dass Nemolizumab als monoklonaler Antikörper in nennenswertem Umfang über die Leber eliminiert wird. Es wurden keine klinischen Studien zur Beurteilung der Wirkung einer Leberfunktionsstörung auf die Pharmakokinetik von Nemolizumab durchgeführt. Eine leichte bis mittelschwere Leberfunktionsstörung beeinflusste die mittels populationspharmakokinetischer Analyse ermittelte PK von Nemolizumab nicht. Für Patienten mit schwerer Leberfunktionsstörung liegen keine Daten vor.
Nierenfunktionsstörung
Es wird nicht erwartet, dass Nemolizumab als monoklonaler Antikörper in nennenswertem Umfang über die Nieren eliminiert wird. Es wurden keine klinischen Studien zur Beurteilung der Wirkung einer Nierenfunktionsstörung auf die Pharmakokinetik von Nemolizumab durchgeführt. In der populationspharmakokinetischen Analyse hatte eine leichte oder mittelschwere Nierenfunktionsstörung keinen klinisch bedeutsamen Einfluss auf die systemische Nemolizumab-Exposition. Für Patienten mit schwerer Nierenfunktionsstörung liegen nur sehr begrenzte Daten vor.
Körpergewicht
Die Nemolizumab-Exposition war bei Patienten mit höherem Körpergewicht geringer.
Atopische Dermatitis
Der durch das Körpergewicht bedingte Unterschied in der systemischen Exposition hatte keine klinisch bedeutsamen Auswirkungen auf die Wirksamkeit. Eine Dosisanpassung aufgrund des Körpergewichts ist nicht erforderlich (siehe Abschnitt 4.2).
Prurigo nodularis
Die Variabilität der systemischen Exposition aufgrund des Körpergewichts hatte eine klinisch bedeutsame Auswirkung auf die Wirksamkeit bei Hautläsionen, beurteilt anhand des IGA-Ansprechens, jedoch nicht auf die Besserung des Pruritus. Eine Dosisanpassung ist daher bei Patienten mit PN erforderlich (siehe Abschnitt 4.2).
Kinder und Jugendliche
Atopische Dermatitis
Aus der populationspharmakokinetischen Analyse ergibt sich kein klinisch relevanter Unterschied in der Pharmakokinetik von Nemolizumab zwischen pädiatrischen Patienten im Alter von 12 bis 17 Jahren und Erwachsenen. Eine Dosisanpassung in dieser Population wird nicht empfohlen.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie und Toxizität bei wiederholter Gabe lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Das mutagene Potenzial von Nemolizumab wurde nicht untersucht; es ist jedoch nicht zu erwarten, dass monoklonale Antikörper die DNA oder Chromosomen verändern.
Mit Nemolizumab wurden keine Studien zur Karzinogenität durchgeführt. Die Auswertung der verfügbaren Erkenntnisse in Bezug auf die IL-31-Hemmung und tierexperimentellen Toxikologiedaten deutet nicht auf ein kanzerogenes Potential hin.
Bei geschlechtsreifen Javaneraffen wurden nach langfristigen subkutanen Behandlungen mit Nemolizumab keine Auswirkungen auf Fruchtbarkeitsparameter beobachtet. In der Gruppe der Muttertiere, die von der frühen Organogenese bis zur Geburt alle zwei Wochen mit 25 mg/kg Nemolizumab behandelt wurden, wurde eine leicht erhöhte Inzidenz von Todesfällen bei den Nachkommen in der frühen postnatalen Phase beobachtet. Die Expositionen der Muttertiere (AUC) waren 43- bzw. 34-mal höher als die beim Menschen empfohlene Höchstdosis bei AD- bzw. PN-Patienten. Ein Zusammenhang zwischen diesem Befund und Nemolizumab kann nicht ausgeschlossen werden.
Pulver zur Herstellung einer Injektionslösung
Saccharose
Trometamol
Trometamolhydrochlorid (zur Einstellung des pH-Werts)
Argininhydrochlorid
Poloxamer 188
Lösungsmittel
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung im Fertigpen
30 Monate
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung in einer Fertigspritze
3 Jahre
Nach erfolgter Rekonstitution muss Nemluvio innerhalb von 4 Stunden verwendet oder entsorgt werden.
Falls erforderlich, kann der Umkarton mit dem Fertigpen oder der Fertigspritze aus dem Kühlschrank genommen und für einen einzelnen Zeitraum von bis zu 90 Tagen bei Raumtemperatur (bis zu 25 °C) aufbewahrt werden. Das Datum der Entnahme aus dem Kühlschrank ist in dem dafür vorgesehenen Feld auf dem Umkarton zu vermerken. Nemluvio darf nicht angewendet werden, wenn das Verfalldatum überschritten ist oder Nemluvio länger als 90 Tage außerhalb des Kühlschranks aufbewahrt wurde (je nachdem, was früher eintritt).
Im Kühlschrank lagern (2 °C – 8 °C).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung im Fertigpen
Einweg-Zweikammer-Ampulle aus Typ-1-Borosilikatglas in einem Autoinjektor mit integrierter („staked“) Edelstahlnadel.
Packungsgröße: 1 Fertigpen, Bündelpackung mit 2 (2 Packungen zu je 1) Fertigpens, Bündelpackung mit 3 (3 Packungen zu je 1) Fertigpens.
Nemluvio 30 mg Pulver und Lösungsmittel zur Herstellung einer Injektionslösung in einer Fertigspritze
Doppelkammer-Einweg-Fertigspritze aus Typ-1-Borosilikatglas, mitgelieferte 27G-Nadel (Edelstahl) mit Schutzabdeckung.
Packungsgröße: 1 Fertigspritze
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Umfassende Anweisungen für die Verabreichung von Nemluvio in einem Fertigpen oder in einer Fertigspritze finden Sie am Ende der Packungsbeilage.
Nemluvio muss 30–45 Min. vor der Rekonstitution aus dem Kühlschrank genommen werden.
Nemluvio muss vor der Rekonstitution einer Sichtprüfung unterzogen werden. Es darf nicht angewendet werden, wenn das Pulver nicht weiß ist oder wenn die Flüssigkeit trüb ist oder Schwebstoffe sichtbar sind. Überprüfen Sie vor der Verabreichung, ob die Lösung klar und farblos bis leicht gelblich ist und keine Schwebstoffe enthält.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Galderma International
La Defense 4, Tour Europlaza
20 Avenue Andre Prothin
92927 Paris La Defense Cedex
Frankreich
EU/1/24/1901/001
EU/1/24/1901/002
EU/1/24/1901/003
EU/1/24/1901/004
Datum der Erteilung der Zulassung: 12. Februar 2025
Februar 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu/ verfügbar.
Verschreibungspflichtig