
▼Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
BEYONTTRA® 356 mg Filmtabletten
Jede Filmtablette enthält Acoramidis-Hydrochlorid, entsprechend 356 mg Acoramidis.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Filmtablette (Tablette).
Weiße, ovale Filmtabletten von ca. 15 mm × 7,5 mm, mit dem BridgeBio-Firmenlogo gefolgt von "ACOR" in schwarzer Farbe auf einer Seite.
BEYONTTRA ist indiziert zur Behandlung der Wildtyp- oder hereditären Transthyretin-Amyloidose bei erwachsenen Patienten mit Kardiomyopathie (ATTR-CM).
Die Therapie sollte durch einen in der Behandlung von Patienten mit Transthyretin-Amyloidose mit Kardiomyopathie (ATTR-CM) erfahrenen Arztes begonnen werden.
Dosierung
Die empfohlene Dosis von Acoramidis beträgt 712 mg (zwei Tabletten zu je 356 mg) oral, zweimal täglich, entsprechend einer täglichen Gesamtdosis von 1 424 mg.
Für Patienten mit New York Heart Association (NYHA) Klasse IV liegen keine Daten zur Wirksamkeit vor (siehe Abschnitt 5.1).
Bei vergessener Einnahme
Es sollte keine doppelte Dosis eingenommen werden, um eine vergessene Einnahme auszugleichen. Die Einnahme sollte zum nächsten geplanten Zeitpunkt fortgesetzt werden.
Besondere Patientengruppen
Ältere Patienten
Bei älteren Patienten (≥ 65 Jahren) ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Nierenfunktionsstörung
Aufgrund der geringen renalen Clearance von Acoramidis ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Für Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min) liegen nur begrenzte Daten vor (siehe Abschnitte 4.4 und 5.2). Es liegen keine Daten für Dialysepatienten vor. Daher sollte Acoramidis bei diesen Patienten mit Vorsicht angewendet werden.
Leberfunktionsstörung
Acoramidis wurde nicht bei Patienten mit Leberfunktionsstörung untersucht und wird daher bei diesen Patienten nicht empfohlen (siehe Abschnitte 4.4 und 5.2).
Kinder und Jugendliche
Es gibt im Anwendungsgebiet „Behandlung der Wildtyp- oder hereditären Transthyretin-Amyloidose mit Kardiomyopathie“ keine relevante Verwendung von Acoramidis bei Kindern und Jugendlichen.
Art der Anwendung
Zum Einnehmen.
Die Filmtabletten müssen im Ganzen geschluckt werden. BEYONTTRA kann mit Wasser und unabhängig von den Mahlzeiten eingenommen werden.
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Leberfunktionsstörung
Acoramidis wurde nicht bei Patienten mit Leberfunktionsstörung untersucht und wird daher bei diesen Patienten nicht empfohlen (siehe Abschnitte 4.2 und 5.2).
Nierenfunktionsstörung
Für Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min) liegen nur begrenzte Daten vor (siehe Abschnitte 4.2 und 5.2). Es liegen keine Daten für Dialysepatienten vor. Daher sollte Acoramidis bei diesen Patienten mit Vorsicht angewendet werden.
Hämodynamische Nierenparameter
Bei mit Acoramidis behandelten Patienten kam es im ersten Behandlungsmonat zu einer anfänglichen Abnahme der geschätzten glomerulären Filtrationsrate (eGFR) und zu einem entsprechenden Anstieg des gemessenen Serumkreatinins (siehe Abschnitt 5.1).
Diese Veränderung der eGFR und des Serumkreatinins war nicht progredient und nicht mit einer Nierenschädigung verbunden. Bei Patienten, deren Behandlung unterbrochen wurde, waren die Veränderungen reversibel, entsprechend einem renalen hämodynamischen Effekt.
Informationen zu sonstigen Bestandteilen
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tablette, d. h. es ist nahezu „natriumfrei“.
Wirkung von Acoramidis auf andere Arzneimittel
Transportsysteme
Basierend auf einer klinischen Studie mit gesunden erwachsenen Probanden ist nicht zu erwarten, dass die Hemmung der organischen Anionentransporter (OAT)-1 und -3 zu klinisch relevanten Arzneimittelwechselwirkungen mit OAT-1- und OAT-3-Substraten führt (z. B. nichtsteroidalen entzündungshemmenden Arzneimitteln, Bumetanid, Furosemid, Lamivudin, Methotrexat, Oseltamivir, Tenofovir, Ganciclovir, Adefovir, Cidofovir, Zidovudin, Zalcitabin).
Basierend auf einer in-vitro-Studie ist in klinisch relevanten Konzentrationen keine Arzneimittelwechselwirkung mit gleichzeitig angewendeten Substraten des Brustkrebs-Resistenz-Proteins (breast cancer resistant protein, BCRP) zu erwarten.
Basierend auf in-vitro-Studien ist es unwahrscheinlich, dass Acoramidis klinisch relevante Uridin-5'-diphospho-(UDP-)Glucuronosyltransferase-abhängige oder Cytochrom-P450-abhängige Wechselwirkungen verursacht. Es konnte jedoch gezeigt werden, dass Acoramidis in vitro CYP2C8 und CYP2C9 inhibiert. Es wurde keine in-vivo-Studie durchgeführt. Daher sollte die gleichzeitige Anwendung von CYP2C8- und CYP2C9-Substraten mit enger therapeutischer Breite mit Vorsicht erfolgen.
Wirkung anderer Arzneimittel auf Acoramidis
Diuretika
Basierend auf einer populationspharmakokinetischen (PK) Analyse hat die gleichzeitige Anwendung von Diuretika bei Patienten keinen Einfluss auf die Steady-State-Plasmakonzentrationen von Acoramidis.
Brustkrebs-Resistenz-Protein-Inhibitoren
Acoramidis ist ein BCRP-Substrat. Basierend auf einer in-vitro-Studie sind keine klinisch relevanten Wechselwirkungen mit BCRP-Inhibitoren zu erwarten.
Arzneimittel zur Reduzierung der Magensäure
Es wurde keine spezielle in-vivo-Interaktionsstudie mit Arzneimitteln zur Reduzierung der Magensäure durchgeführt. Daher ist der Einfluss dieser Arzneimittel auf die Pharmakokinetik von Acoramidis unbekannt. Trotz der ausgeprägten pH-abhängigen Löslichkeit von Acoramidis im physiologischen pH-Bereich, wurden in der Phase-3-Studie keine Unterschiede in der systemischen Acoramidis-Exposition oder im pharmakodynamischen Marker (TTR-Stabilisierung) zwischen Patienten, die Magensäure-reduzierende Arzneimittel einnahmen, und Patienten, die keine Magensäure-reduzierenden Arzneimittel einnahmen, beobachtet.
Auswirkung auf Laboruntersuchungen
Acoramidis kann die Serumkonzentrationen des freien Thyroxins verringern, ohne gleichzeitige Veränderung des Thyreotropins (thyroid stimulating hormone, TSH). Klinische Befunde, die auf eine Schilddrüsenfunktionsstörung hindeuten, wurden nicht beobachtet.
Schwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Acoramidis bei Schwangeren vor.
Tierexperimentelle Studien haben eine Entwicklungstoxizität bei einer Dosis gezeigt, die auch eine maternale Toxizität verursachte (siehe Abschnitt 5.3). Die Anwendung von Acoramidis während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die nicht verhüten, wird nicht empfohlen.
Stillzeit
Es ist nicht bekannt ob Acoramidis oder seine Metaboliten in die Muttermilch übergehen. Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden (siehe Abschnitt 5.3). Acoramidis soll während der Stillzeit nicht angewendet werden.
Fertilität
Es liegen keine Daten zur Auswirkung auf die menschliche Fertilität vor. Eine Beeinträchtigung der Fertilität wurde in präklinischen Studien bei supratherapeutischen Expositionen nicht beobachtet.
BEYONTTRA hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
In der klinischen Studie waren die am häufigsten berichteten Nebenwirkungen Durchfall (11,6 %) und Gicht (11,2 %).
Tabellarische Auflistung der Nebenwirkungen
Die Sicherheitsdaten spiegeln die Exposition von 421 Patienten mit ATTR-CM gegenüber zweimal täglicher oraler Einnahme von 712 mg Acoramidis (zwei Tabletten zu je 356 mg) in einer randomisierten, doppelblinden, placebokontrollierten Phase-3-Studie mit einer festgelegten Behandlungsdauer von 30 Monaten wider.
Die Nebenwirkungen sind nachstehend nach Systemorganklasse (SOC) gemäß MedDRA und Häufigkeitskategorien unter Verwendung der Standardkonvention aufgeführt: Sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10) und gelegentlich (≥ 1/1 000, < 1/100). Die in der nachstehenden Tabelle aufgeführten Nebenwirkungen stammen aus kumulativen klinischen Daten von Teilnehmern mit ATTR-CM.
Tabelle 1: Auflistung der Nebenwirkungen
Systemorganklasse | Sehr häufig |
Erkrankungen des Gastrointestinaltrakts | Diarrhö |
Stoffwechsel- und Ernährungsstörungen | Gicht |
Beschreibung ausgewählter Nebenwirkungen
Die Mehrzahl der Fälle von Durchfall und Gicht waren nicht schwerwiegend und klangen wieder ab.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das Bundesinstitut für Arzneimittel und Medizinprodukte, Abt. Pharmakovigilanz, Kurt-
Georg-Kiesinger-Allee 3, D-53175 Bonn, Website: http://www.bfarm.de anzuzeigen.
Es liegen keine klinischen Erfahrungen mit Überdosierungen vor.
Im Verdachtsfall einer Überdosis sollte eine symptomatische und unterstützende Behandlung erfolgen.
Pharmakotherapeutische Gruppe: Herztherapie, sonstige kardiale Präparate. ATC-Code: C01EB25.
Wirkmechanismus
Die Transthyretin-Amyloidose mit Kardiomyopathie wird durch die Dissoziation des Transthyretin-(TTR-)Tetramers in seine Monomere ausgelöst. Durch Fehlfaltung aggregieren diese zu oligomeren Amyloid-Vorläufern, die sich im Herzen ablagern, wo sie sich zu Amyloidfibrillen organisieren.
Acoramidis ist ein spezifischer TTR-Stabilisator. Acoramidis bildet Wasserstoffbrückenbindungen mit benachbarten Serinresten innerhalb beider Thyroxin-Bindungsstellen des Tetramers und ahmt damit die Wirkweise der protektiven T119M Variante nach. Diese Wechselwirkung erhöht die Stabilität des Tetramers, hemmt seine Dissoziation in Monomere und verlangsamt so den amyloidogenen Prozess, der zu ATTR-CM führt.
Pharmakodynamische Wirkungen
Unter Acoramidis wurde eine nahezu vollständige Transthyretin-Stabilisierung beim Wildtyp und bei allen getesteten amyloidogenen Genotypvarianten beobachtet, einschließlich der am weitesten verbreiteten Genotypen V30M (p.V50M), T60A (p.T80A) und V122I (p.V142I). In der Studie ATTRibute‑CM wurde bei Patienten (Wildtyp- und hereditäre ATTR), die mit Acoramidis (zweimal täglich 712 mg) behandelt wurden, bei der ersten Messung nach Behandlungsbeginn (Tag 28) eine nahezu vollständige (≥ 90 %) TTR-Stabilisierung beobachtet, die über den gesamten Beobachtungszeitraum von 30 Monaten aufrechterhalten wurde. Bei allen Messungen nach Behandlungsbeginn (von Tag 28 bis Monat 30) war der TTR-Spiegel in der Acoramidis-Gruppe höher als in der Placebo-Gruppe (in Monat 30 mittlere Veränderung gegenüber dem Ausgangswert 9,1 mg/dl unter Acoramidis gegenüber 1,3 mg/dl unter Placebo).
In der Studie ATTRibute-CM fiel der Anstieg des N‑terminalen Prohormons des natriuretischen Peptids Typ B (NT-proBNP) zugunsten von Acoramidis im Vergleich zu Placebo aus und betrug in Monat 30 etwa die Hälfte. Darüber hinaus wurde ein geringerer Anstieg von Troponin I unter Acoramidis im Vergleich zu Placebo beobachtet.
In der Studie ATTRibute-CM betrug das mittlere Serumkreatinin (und die eGFR) zu Studienbeginn 110,0 μmol/l (eGFR: 60,9 ml/min/1,73 m2) in der Acoramidis-Gruppe und 109,0 μmol/l (eGFR: 61,0 ml/min/1,73 m2) in der Placebogruppe. An Tag 28 wurde eine Veränderung des mittleren Serumkreatinins (und der eGFR) gegenüber dem Ausgangswert beobachtet, die in der Acoramidis-Gruppe größer war (an Tag 28 beobachtete Serumkreatinin-Werte: 129,3 μmol/l, eGFR: 52,4 ml/min/1,73 m2) als in der Placebogruppe (an Tag 28 beobachtete Serumkreatinin-Werte: 110,6 μmol/l, eGFR: 60,0 ml/min/1,73 m2). Nach Tag 28 blieb das Serumkreatinin (eGFR) in der Acoramidis-Gruppe für den Rest der Studie stabil. In der Placebogruppe wurde ein progressiver Anstieg des Serumkreatinins und eine entsprechende progressive Abnahme der eGFR vom Ausgangswert bis Monat 30 beobachtet. In Monat 30 betrug das Serumkreatinin für Acoramidis 123,4 μmol/l (eGFR: 55,1 ml/min/1,73 m2) bzw. für Placebo 117,2 μmol/l (eGFR: 57,2 ml/min/1,73 m2). Der beobachtete Anstieg des Serumkreatinins und die entsprechende Abnahme der eGFR, die bei mit Acoramidis behandelten Patienten beobachtet wurden, waren bei Unterbrechung der Therapie reversibel.
Kardiale Elektrophysiologie
Die höchste an gesunden erwachsenen Probanden untersuchte Einzeldosis von Acoramidis in Höhe von 1 780 mg hatte keinen klinisch relevanten Effekt auf die kardiale Leitung oder Repolarisation (es wurde kein Konzentrations-QTc-Effekt beobachtet). Diese Beobachtungen deuten auf ein geringes Risiko für Proarrhythmie hin.
Klinische Wirksamkeit
ATTRibute-CM war eine multizentrische, internationale, randomisierte, doppelblinde, placebokontrollierte klinische Studie, die an 632 Teilnehmern mit Wildtyp- oder hereditärer ATTR-CM (umfasst hereditär und de novo) und Herzinsuffizienz der NYHA-Klasse I‑III, mit derzeit bestehenden oder früheren Symptomen von Herzinsuffizienz, durchgeführt wurde. Die Teilnehmer wurden im Verhältnis 2:1 randomisiert und erhielten 30 Monate lang zweimal täglich 712 mg Acoramidis (n = 421) oder ein entsprechendes Placebo (n = 211). Der Einschluss in die Behandlungsgruppen wurde nach Vorliegen einer hereditären ATTR-CM (ATTRv-CM) oder einer Wildtyp-ATTR-CM (ATTRwt-CM), sowie nach dem Schweregrad der Erkrankung, d. h. dem NT-proBNP-Spiegel und der Nierenfunktion, definiert durch die eGFR, stratifiziert. Patienten mit einer eGFR < 15 ml/min/1,73 m2 wurden von der Teilnahme an der Studie ausgeschlossen.
Tabelle 2: Demografie und Baseline-Charakteristika (mITT-Population1)
Merkmal | Acoramidis | Placebo |
Alter – Jahre | ||
Mittelwert (Standardabweichung) | 77,3 (6,5) | 77,0 (6,7) |
Geschlecht – Anzahl (%) | ||
Männlich | 374 (91,4) | 181 (89,6) |
Weiblich | 35 (8,6) | 21 (10,4) |
TTR-Genotyp2 – Anzahl (%) | ||
ATTRv | 39 (9,5) | 20 (9,9) |
ATTRwt | 370 (90,5) | 182 (90,1) |
NYHA-Klasse – Anzahl (%) | ||
NYHA Klasse I | 51 (12,5) | 17 (8,4) |
NYHA Klasse II | 288 (70,4) | 156 (77,2) |
NYHA Klasse III | 70 (17,1) | 29 (14,4) |
eGFR2 (ml/min/1,73 m2) – Anzahl (%) | ||
eGFR ≥ 45 | 344 (84,1) | 173 (85,6) |
eGFR < 45 | 65 (15,9) | 29 (14,4) |
NT-proBNP2 (pg/ml) – Anzahl (%) | ||
≤ 3 000 | 268 (65,5) | 133 (65,8) |
> 3 000 | 141 (34,5) | 69 (34,2) |
ATTR-NAC-Stadium3 – Anzahl (%) | ||
I | 241 (58,9) | 120 (59,4) |
II | 130 (31,8) | 66 (32,7) |
III | 38 (9,3) | 16 (7,9) |
Permanenter Herzschrittmacher in der Anamnese – Anzahl (%) | 77 (18,8) | 38 (18,8) |
Vorhofflimmern in der Anamnese – Anzahl (%) | 236 (57,7) | 117 (57,9) |
Abkürzungen: ATTRv = hereditäre Transthyretin-Amyloidose, ATTRwt = Wildtyp-Transthyretin-Amyloidose, NAC = National Amyloidosis Centre (London, Vereinigtes Königreich), NYHA = New York Heart Association, eGFR = geschätzte glomeruläre Filtrationsrate, NT-proBNP = N-terminales Prohormon des natriuretischen Peptids Typ B, TTR = Transthyretin
1 mITT = modifizierte Intent-to-Treat (Baseline-eGFR ≥ 30 ml/min/1,73 m2).
2 Stratifizierungsfaktoren.
3 NAC-Stadium I (NT-proBNP ≤ 3 000 pg/ml und eGFR ≥ 45 ml/min/1,73 m2), Stadium II (NT-proBNP ≤ 3 000 pg/ml und eGFR < 45 ml/min/1,73 m2 oder NT proBNP > 3 000 pg/ml und eGFR ≥ 45 ml/min/1,73 m2), Stadium III (NT-proBNP > 3 000 pg/ml und eGFR < 45 ml/min/1,73 m2).
Die Teilnehmer durften nach 12 Monaten in der Studie mit der offenen Einnahme von Tafamidis beginnen, wenn dieses als Begleitmedikation verschrieben wurde. Insgesamt erhielten 107 Teilnehmer Tafamidis, 61 (14,9 %) im Acoramidis-Studienarm und 46 (22,8 %) im Placebo-Studienarm.
Das primäre Ziel der Studie war der Nachweis der Überlegenheit von Acoramidis gegenüber Placebo in Bezug auf einen hierarchischen Endpunkt, der die Gesamtmortalität und die kumulative Häufigkeit kardiovaskulär bedingter Hospitalisierungen umfasste. Zu den sekundären Zielen gehörten die Bewertung der Gesamtmortalität, der kardiovaskulär bedingten Hospitalisierungen, des 6-Minuten-Gehtests, des Kansas City Cardiomyopathy Questionnaire (KCCQ)-Overall Summary Scores (ein Maß für die Lebensqualität), des Serum-TTR-Spiegels und des NT‑proBNP. Die wesentlichen Wirksamkeitsanalysen wurden bei den 611 Teilnehmern der modifizierten Intent-to-Treat-(mITT‑)Population ohne Adjustierungen nach der unverblindeten Anwendung von Tafamidis durchgeführt.
Wirksamkeitsanalyse
Die Wirksamkeitsanalyse wandte den stratifizierten Finkelstein-Schoenfeld (F-S)-Test hierarchisch auf Gesamtmortalität und die kumulative Häufigkeit kardiovaskulär bedingter Hospitalisierungen über die 30-monatige Studie an. Die Methode verglich jeden Patienten mit jedem anderen Patienten in jedem Stratum; dabei wurde paarweise auf hierarchische Weise vorgegangen, wobei zuerst die Gesamtmortalität gefolgt von der Häufigkeit der kardiovaskulär bedingten Hospitalisierungen, wenn eine Differenzierung der Patienten basierend auf Gesamtmortalität nicht möglich war, herangezogen wurde. Das Ergebnis dieser Analyse war statistisch signifikant (Tabelle 3).
Die Gesamtmortalität betrug 19,3 % bzw. 25,7 % in der Acoramidis- bzw. Placebogruppe. Die Mehrzahl (79 %) der Todesfälle war kardiovaskulär bedingt, wobei Acoramidis eine relative Risikoreduktion der kardiovaskulären Mortalität um 30 % im Vergleich zu Placebo zeigte. Kardiovaskuläre Mortalität wurde bei 14,9 % bzw. 21,3 % der Teilnehmer in der Acoramidis- bzw. Placebo-Gruppe berichtet; Hazard Ratio: 0,709 (95 % KI: 0,476; 1,054; p = 0,0889; Cox-Regressionsmodell).
Eine Cox-Regressionsanalyse zeigte eine 35,5%ige Abnahme des Risikos für die Kombination aus Gesamtmortalität oder erstmaliger kardiovaskulär bedingter Hospitalisierung (Hazard Ratio: 0,645 [95 % KI: 0,500; 0,832; p = 0,0008]). Die Kaplan-Meier-Kurven trennten sich ab Monat 3 und gingen danach bis Monat 30 stetig auseinander (Abbildung 1).
Die in der mITT-Population gezeigten Ergebnisse der Wirksamkeitsanalyse in Bezug auf Gesamtmortalität und kardiovaskulär bedingte Hospitalisierungen wurden auch in der ITT-Population beobachtet (alle randomisierten Probanden unabhängig von Baseline eGFR).
Tabelle 3: Ergebnisse der Wirksamkeitsanalyse nach Finkelstein-Schoenfeld, Gesamtmortalität und kardiovaskulär bedingte Hospitalisierung bis Monat 30 in ATTRibute-CM (mITT-Population)
Parameter | Acoramidis | Placebo |
Kombination von ACM und kumulativer Häufigkeit von CVH | ||
Win-Ratio (95%‑KI) | 1,464 (1,067; 2,009) | |
F‑S1 p‑Wert | p = 0,0182 | |
Anzahl (%) der Patienten, die in Monat 30 noch lebten2 | 330 (80,7 %) | 150 (74,3 %) |
Anzahl (%) der Patienten mit CVH | 109 (26,7 %) | 86 (42,6 %) |
Relatives Risiko4 | 0,496 | |
Abkürzungen: F-S = Finkelstein-Schoenfeld; ACM = Gesamtmortalität; CVH = kardiovaskulär bedingte Hospitalisierung; mITT = modifizierte Intent-to-Treat; KI = Konfidenzintervall
1 Die F-S-Methode vergleicht jeden Patienten mit jedem anderen Patienten in jedem Stratum in einer hierarchischen Weise, beginnend mit der Gesamtmortalität. Wenn eine Differenzierung basierend auf der Gesamtmortalität nicht möglich war, erfolgte die Differenzierung basierend auf der Häufigkeit der kardiovaskulär bedingten Hospitalisierungen.
2 Herztransplantationen und Implantationen von Systemen zur mechanischen Unterstützung der Herzfunktion werden als Anzeichen für ein nahendes Endstadium angesehen. Aus diesem Grund werden diese Ereignisse in der Analyse mit Todesfällen gleichgestellt. Daher werden solche Patienten nicht in die Zählung der „Anzahl der Patienten, die in Monat 30 noch lebten” einbezogen, selbst wenn die Patienten basierend auf der Folgeuntersuchung des Vitalstatus in Monat 30 noch am Leben sind. Der Vitalstatus in Monat 30 war bei allen Patienten bekannt.
3 Die Häufigkeit von kardiovaskulär bedingten Hospitalisierungen pro Patient und Jahr wird wie folgt berechnet: (Anzahl der gesamten kardiovaskulär bedingten Hospitalisierungen pro Patient)/(Dauer der Nachbeobachtung in Jahren) und umfasst Ereignisse von klinischem Interesse (events of clinical interest, EOCI). EOCI ist definiert als Arztbesuche (z. B. Notaufnahme, Notfallambulanz, Tagesklinik) von < 24 Stunden für eine intravenöse Diuretikatherapie zur Behandlung einer dekompensierten Herzinsuffizienz.
4 Aus dem negativen binomialen Regressionsmodell.
Abbildung 1: Zeit bis zum Tod jeglicher Ursache oder der ersten kardiovaskulär bedingten Hospitalisierung
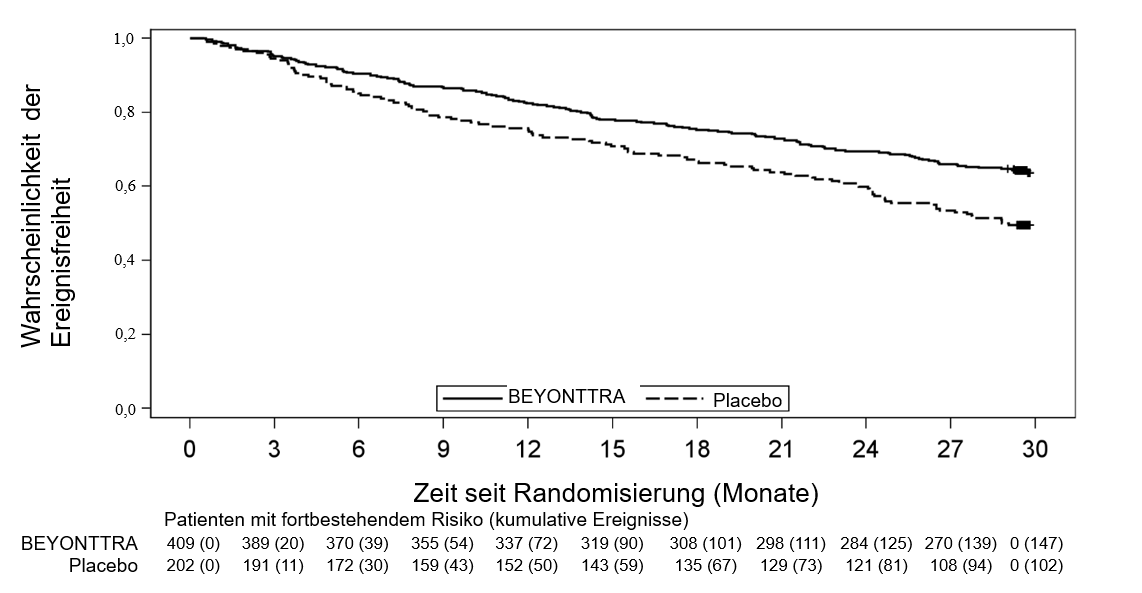
Distanz im 6-Minuten-Gehtest (6MWD) und KCCQ
Der Behandlungseffekt von Acoramidis auf die Leistungsfähigkeit und den Gesundheitszustand wurde durch die 6MWD und mithilfe des KCCQ Overall Summary Score (KCCQ-OS; bestehend aus den Domänen körperliche Beeinträchtigung, Symptome, soziale Beeinträchtigung und Lebensqualität) beurteilt (Tabelle 4). Ein Behandlungseffekt zugunsten von Acoramidis wurde für die 6MWD und den KCCQ-OS erstmals in Monat 18 bzw. Monat 3 beobachtet und hielt bis Monat 30 an.
Tabelle 4: Ergebnisse von 6MWD und KCCQ-OS
Endpunkte* | Baseline-Mittelwert | Veränderung von Baseline bis Monat 30, KQ-Mittelwert (SE) | Differenz KQ-Mittelwert Behandlung gegen Placebo (96 % KI) | p-Wert | ||
Acoramidis | Placebo | Acoramidis | Placebo | |||
6MWD (Meter) | 362,78 | 351,51 | ‑64,65 | ‑104,29 | 39,64 | < 0,0001 |
KCCQ-OS | 71,73 | 70,48 | ‑11,48 | ‑21,42 | 9,94 | < 0,0001 |
Abkürzungen: 6MWD = 6-Minuten-Gehtest; KI = Konfidenzintervall; KCCQ-OS = Kansas City Cardiomyopathy Questionnaire Overall Summary Score, KQ = kleinste Quadrate, SD = Standardabweichung, SE = Standardfehler
* Höhere Werte weisen auf einen besseren Gesundheitszustand hin.
Subgruppenanalyse
Die Ergebnisse des F-S-Tests, angewendet auf Gesamtmortalität und kardiovaskulär bedingte Hospitalisierungen (ergänzt durch die Win-Ratio), fielen über alle Stratifizierungsparameter (Wildtyp oder hereditär), NYHA-Klasse und ATTR-Stadium nach National Amyloidosis Centre (NAC) im Vergleich zu Placebo einheitlich zugunsten von Acoramidis aus (Abbildung 2).
Abbildung 2: Hierarchische Kombination aus Gesamtmortalität und kardiovaskulär bedingter Hospitalisierung, Finkelstein-Schoenfeld- und Win-Ratio-Ergebnisse insgesamt und nach Subgruppe (mITT-Population)1
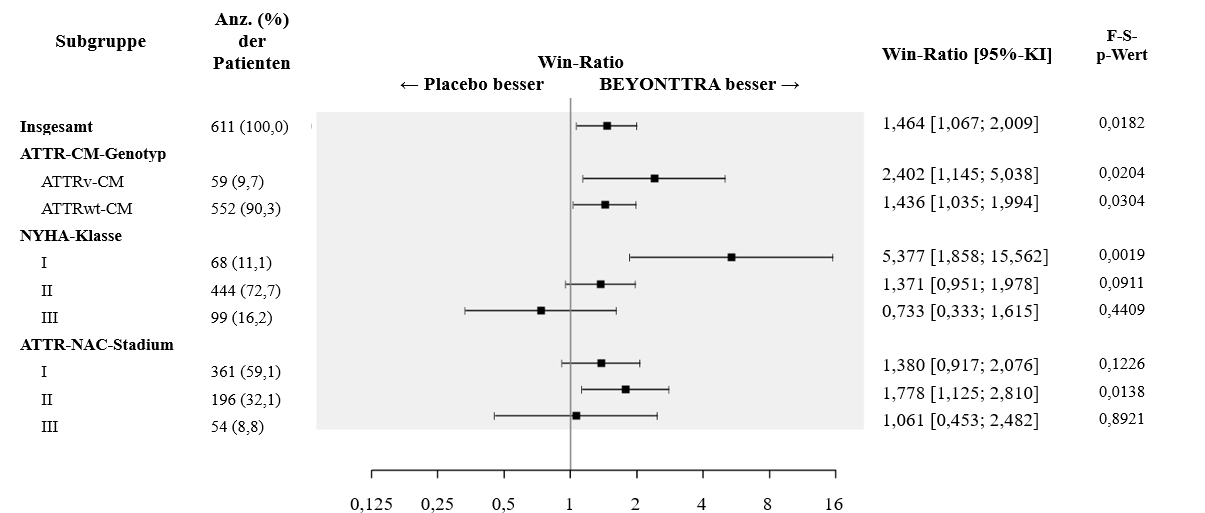
Abkürzungen: ATTRwt-CM = Wildtyp ATTR-CM; ATTRv-CM = hereditäre ATTR-CM; F-S = Finkelstein-Schoenfeld; NAC = National Amyloidosis Centre (London, Vereinigtes Königreich); NYHA = New York Heart Association; NAC-Stadium I (NT-proBNP ≤ 3 000 pg/ml und eGFR ≥ 45 ml/min/1,73 m2), Stadium II (NT-proBNP ≤ 3 000 pg/ml und eGFR < 45 ml/min/1,73 m2 oder NT proBNP > 3 000 pg/ml und eGFR ≥ 45 ml/min/1,73 m2), Stadium III (NT-proBNP > 3 000 pg/ml und eGFR < 45 ml/min/1,73 m2)
1 Die Win-Ratio ist die Anzahl der Paare mit „Gewinnen“ des Acoramidis-behandelten Patienten dividiert durch die Anzahl der Paare mit „Gewinnen“ des Placebo-behandelten Patienten.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat für BEYONTTRA eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen in ATTR-CM gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
Resorption
Der Anstieg der Expositionsparameter (Fläche unter der Konzentrations-Zeit-Kurve [AUC] und maximale Plasmakonzentration [Cmax]) war bei einmaliger (bis zu 1 780 mg) oder mehrfacher (bis zu 712 mg) zweimal täglicher Einnahme unterproportional zur Dosis.
Nach oraler Anwendung wird Acoramidis schnell resorbiert und die maximale Plasmakonzentration von unverändertem Acoramidis in der Regel innerhalb von 1 Stunde erreicht. Bei Acoramidis-Dosierungen von 44,5 mg einmal täglich bis 712 mg einmal täglich wurde ein Anstieg der Plasmakonzentration beobachtet. Die Plasmaexposition erreichte bei Acoramidis-Dosierungen von 712 mg bis 1 068 mg eine Sättigung. Ein Steady-State-Zustand wird nach 10-tägiger Einnahme von 712 mg zweimal täglich erreicht, und eine wiederholte Einnahme führt zu einer geringen (etwa 1,3- bis 1,6‑fachen) Anreicherung von Acoramidis.
Die absolute Bioverfügbarkeit ist nicht bekannt; mindestens 75-80 % der oral angewendeten Einzeldosis von 712 mg werden jedoch auf der Grundlage einer humanen ADME-Studie (Absorption, Verteilung, Metabolismus, Ausscheidung) resorbiert.
Das Gesamtausmaß der Resorption von Acoramidis wird nicht durch Nahrungsaufnahme beeinflusst.
Verteilung
Das scheinbare Steady-State-Verteilungsvolumen beträgt bei zweimal täglicher Einnahme von 712 mg Acoramidis 654 Liter. Die in-vitro-Bindung von Acoramidis an humane Plasmaproteine beträgt 96,4 %. Acoramidis bindet primär an TTR.
Biotransformation
Die Metabolisierung von Acoramidis wurde nach Anwendung einer oralen Einzeldosis von [14C]-Acoramidis bei gesunden erwachsenen Probanden charakterisiert. Acoramidis wird überwiegend durch Glucuronidierung metabolisiert, wobei Acoramidis-β-D-Glucuronid (Acoramidis-AG) der vorherrschende Metabolit ist (7,6 % der gesamten zirkulierenden Radioaktivität). Acoramidis-AG weist eine etwa 3‑fach geringere pharmakologische Aktivität auf als Acoramidis, hat ein geringes Potenzial für kovalente Bindungen und trägt nicht wesentlich zur pharmakologischen Aktivität bei.
Elimination
Die terminale Halbwertszeit von Acoramidis beträgt etwa 27 Stunden nach einer Einzeldosis. Im Steady-State-Zustand beträgt die scheinbare orale Clearance von Acoramidis 15,6 l/h.
Nach Anwendung einer oralen Einzeldosis von [14C]-Acoramidis bei gesunden erwachsenen Probanden wurden etwa 34 % der Dosis-Radioaktivität im Stuhl (mit Acoramidis als Hauptkomponente) und etwa 68 % im Urin wiedergefunden. Der Anteil an unverändertem Acoramidis im Urin lag bei < 10 %.
Besondere Patientengruppen
Es wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik von Acoramidis aufgrund des Alters (18,0-89,3 Jahre), der ethnischen Zugehörigkeit (einschließlich japanisch und nicht-japanisch), des Geschlechts oder einer eingeschränkten Nierenfunktion (eGFR 25,4-157 ml/min/1,73 m2) beobachtet.
Basierend auf dem Populations-PK-Modell war die AUC von Acoramidis im Steady-State-Zustand bei gesunden Probanden um 37 % höher als bei der Patientenpopulation. Im Vergleich zu weißen Probanden war die AUC im Steady-State-Zustand bei schwarzen Probanden um 23 % höher und bei nicht-weißen, nicht-schwarzen Probanden um 38 % höher. Diese Effekte liegen im Bereich der interindividuellen Variabilität (CV = 38 %). Das Modell sagte auch das Fehlen klinisch signifikanter Unterschiede in der Pharmakokinetik von Acoramidis aufgrund des Körpergewichts im Bereich von 50,9 bis 133 kg voraus.
Eine speziell angelegte Studie zu Nierenfunktionsstörungen wurde nicht durchgeführt, da Acoramidis nicht wesentlich über die Nieren eliminiert wird und der Hauptmetabolit (Acoramidis-AG) keinen klinisch relevanten Beitrag zur pharmakologischen Aktivität in der untersuchten Population leistet. Daten für Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min) sind begrenzt, und es liegen keine Daten für Dialysepatienten vor. Die Clearance des Acoramidis-Metaboliten Acoramidis-AG könnte durch eine schwere Nierenfunktionsstörung beeinträchtigt werden, was möglicherweise zu einer höheren systemischen Exposition von Acoramidis-AG führt. Obwohl nicht zu erwarten ist, dass dieser potenzielle Anstieg der Acoramidis-AG-Exposition einen klinisch relevanten Beitrag zur pharmakologischen Aktivität leistet, sollte Acoramidis bei Patienten mit schweren Nierenfunktionsstörungen mit Vorsicht angewendet werden.
Acoramidis wurde nicht bei Patienten mit Leberfunktionsstörung untersucht.
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität, zum kanzerogenen Potential sowie zur Entwicklungs- und Reproduktionstoxizität (Fertilität und embryofötale Entwicklung), lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
In der Studie zur prä- und postnatalen Entwicklung bei Ratten wurden nach Gabe von Acoramidis (1 000 mg/kg/Tag) an das Muttertier während der Gestation und Laktationszeit eine verringerte Überlebensrate der Jungtiere, ein reduziertes Gewicht der Jungtiere und Lerndefizite beobachtet. Bei dieser Dosis wurde auch eine schwere maternale Toxizität einschließlich Mortalität und Gewichtsverlust während der Organogenese beobachtet. Der NOAEL-Wert (No-Observed-Adverse-Effect-Level) in prä- und postnatalen Entwicklungstoxizitätsstudien an Ratten wurde bei der getesteten Acoramidis-Dosierung von 350 mg/kg/Tag ermittelt (die AUC-Werte betrugen das annähernd 21‑fache der Exposition beim Menschen bei der klinischen Dosis von Acoramidis).
Es wurden keine tierexperimentellen Studien zur Plazentagängigkeit und zur Milchexkretion durchgeführt.
Tablettenkern
Mikrokristalline Cellulose (E 460)
Croscarmellose-Natrium (E 468)
Kolloidale hydratisierte Kieselsäure (E 551)
Magnesiumstearat (E 470b)
Filmüberzug
Polyvinylalkohol-polyethylenglycol-Propfcopolymer (E 1209)
Talkum (E 553b)
Titandioxid (E 171)
Glycerylmonocaprylocaprat, Typ I (E 471)
Polyvinylalkohol (E 1203)
Druckfarbe
Schwarzes Eisenoxid (E 172)
Propylenglycol (E 1520)
Hypromellose 2910 (E 464)
Nicht zutreffend.
30 Monate
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Blisterpackungen – Thermogeformte Blister mit zwei Kavitäten aus PVC/PCTFE mit einer Aluminium-Deckfolie
Packungsgrößen: 120 Tabletten in 6 Blisterstreifen mit je 10 Kavitäten (2 Tabletten pro Kavität)
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Bayer AG
51368 Leverkusen
Deutschland
EU/1/24/1906/001
Datum der Erteilung der Zulassung: 10. Februar 2025
Juni 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur verfügbar: https://www.ema.europa.eu.
-------------------------------------------------------------------------------------------------------------------------------
Verschreibungspflichtig
Bayer Vital GmbH
51368 Leverkusen
Tel.: +49 (0)214-30 513 48
Fax: +49 (0)214-2605 516 03
E-Mail: medical-information@bayer.com
DE/2